School of Computer Science and Engineering, Pusan National University, Busan, Republic of Korea.
Research & Development, NuclixBio, Seoul, Republic of Korea.
PLoS One. 2021 Jun 25;16(6):e0253760. doi: 10.1371/journal.pone.0253760. eCollection 2021.
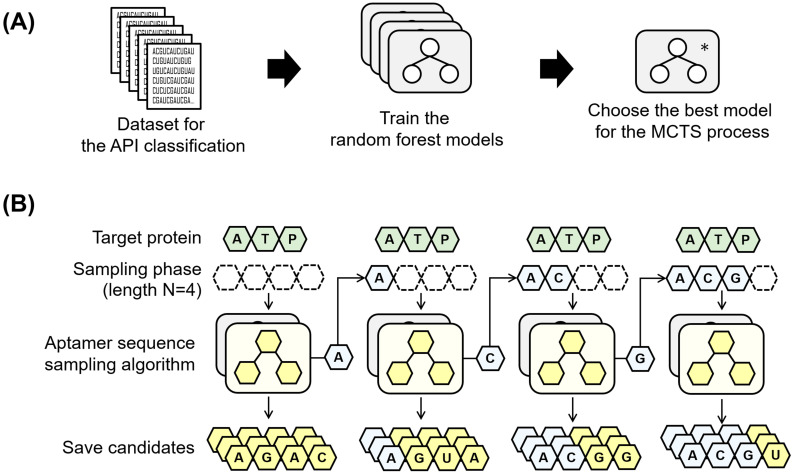
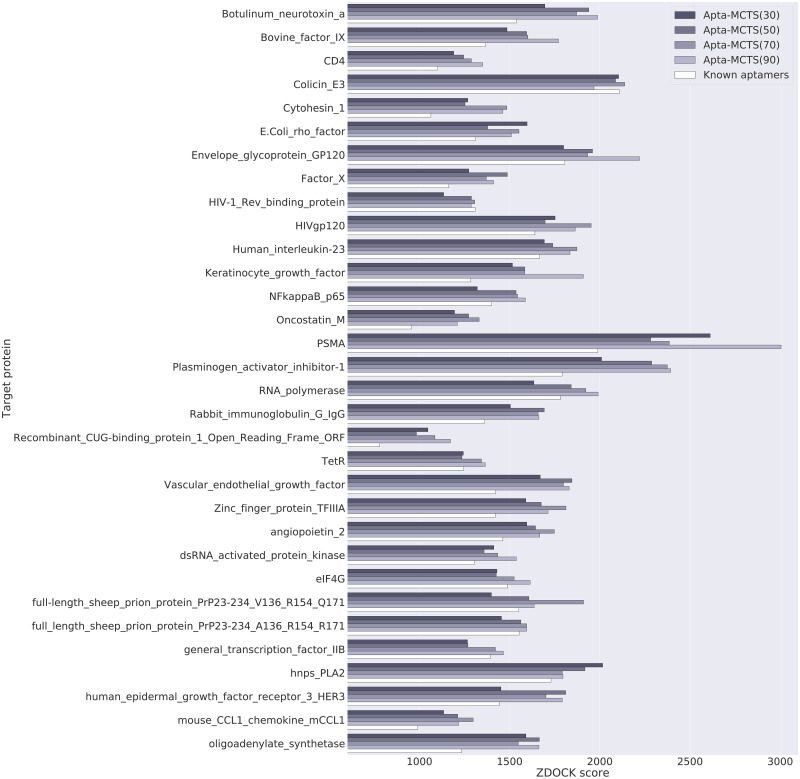
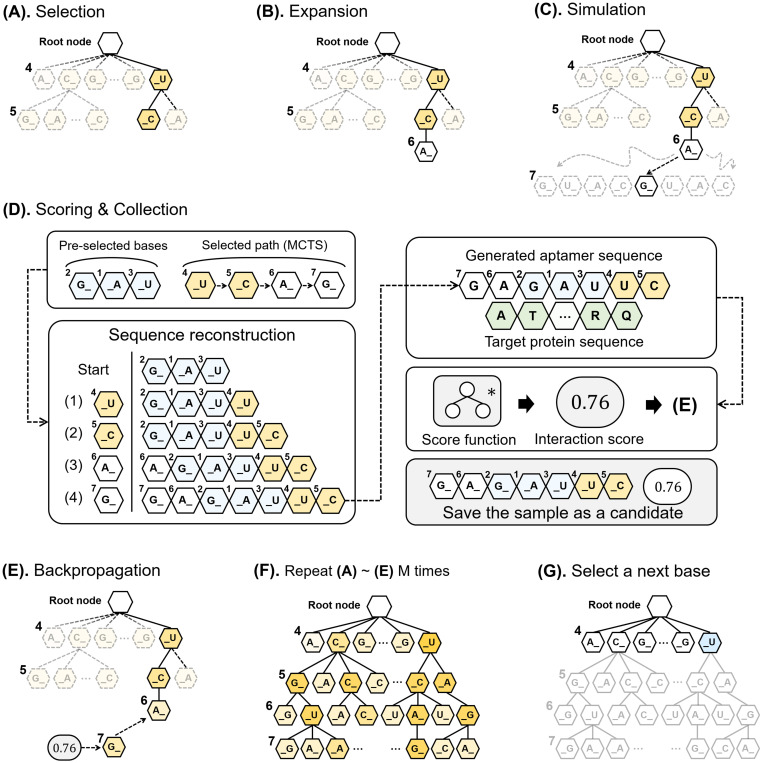
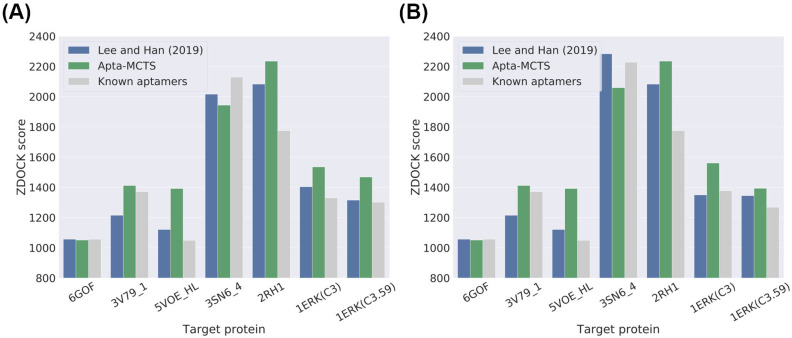
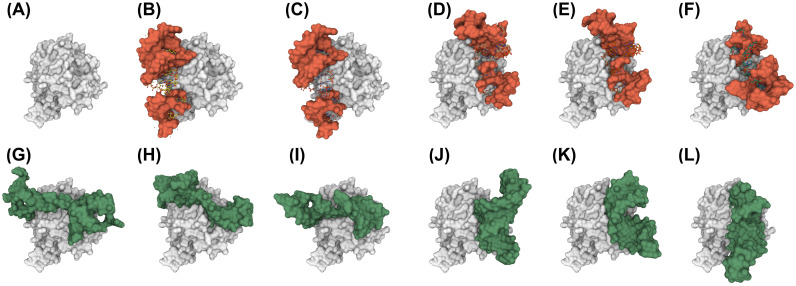
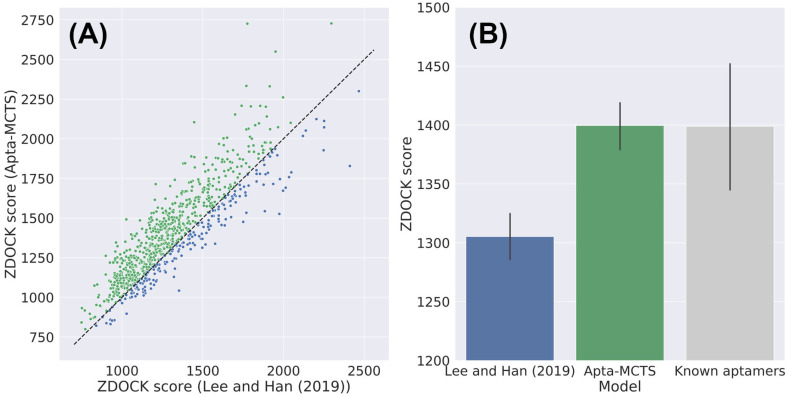
Oligonucleotide-based aptamers, which have a three-dimensional structure with a single-stranded fragment, feature various characteristics with respect to size, toxicity, and permeability. Accordingly, aptamers are advantageous in terms of diagnosis and treatment and are materials that can be produced through relatively simple experiments. Systematic evolution of ligands by exponential enrichment (SELEX) is one of the most widely used experimental methods for generating aptamers; however, it is highly expensive and time-consuming. To reduce the related costs, recent studies have used in silico approaches, such as aptamer-protein interaction (API) classifiers that use sequence patterns to determine the binding affinity between RNA aptamers and proteins. Some of these methods generate candidate RNA aptamer sequences that bind to a target protein, but they are limited to producing candidates of a specific size. In this study, we present a machine learning approach for selecting candidate sequences of various sizes that have a high binding affinity for a specific sequence of a target protein. We applied the Monte Carlo tree search (MCTS) algorithm for generating the candidate sequences using a score function based on an API classifier. The tree structure that we designed with MCTS enables nucleotide sequence sampling, and the obtained sequences are potential aptamer candidates. We performed a quality assessment using the scores of docking simulations. Our validation datasets revealed that our model showed similar or better docking scores in ZDOCK docking simulations than the known aptamers. We expect that our method, which is size-independent and easy to use, can provide insights into searching for an appropriate aptamer sequence for a target protein during the simulation step of SELEX.
寡核苷酸适体是一种具有单链片段的三维结构,具有大小、毒性和通透性等各种特性。因此,适体在诊断和治疗方面具有优势,并且是可以通过相对简单的实验生产的材料。指数富集的配体系统进化(SELEX)是生成适体最广泛使用的实验方法之一;然而,它非常昂贵且耗时。为了降低相关成本,最近的研究使用了计算方法,例如基于序列模式的适体-蛋白相互作用(API)分类器,用于确定 RNA 适体与蛋白质之间的结合亲和力。其中一些方法生成与靶蛋白结合的候选 RNA 适体序列,但它们仅限于产生特定大小的候选物。在这项研究中,我们提出了一种机器学习方法,用于选择具有高结合亲和力的各种大小的候选序列特定靶蛋白的特定序列。我们使用基于 API 分类器的评分函数应用蒙特卡罗树搜索(MCTS)算法生成候选序列。我们设计的 MCTS 树结构允许核苷酸序列采样,并且获得的序列是潜在的适体候选物。我们使用对接模拟的分数进行了质量评估。我们的验证数据集表明,与已知的适体相比,我们的模型在 ZDOCK 对接模拟中显示出相似或更好的对接分数。我们期望我们的方法,它是独立于大小且易于使用的,可以在 SELEX 的模拟步骤中为靶蛋白搜索合适的适体序列提供见解。