Shanghai Zerun Biotechnology Co., Ltd., Pilot Department, Building 9, 1690 Zhangheng Road Pudong, Shanghai, 201203, China.
BMC Cancer. 2021 Jun 26;21(1):733. doi: 10.1186/s12885-021-08412-4.
This study aimed to explore and identify key genes and signaling pathways that contribute to the progression of cervical cancer to improve prognosis.
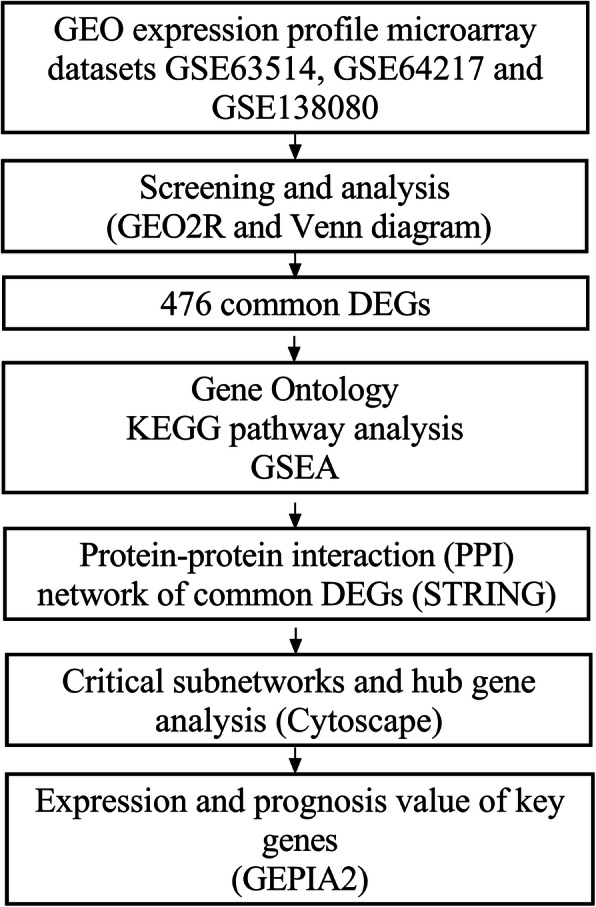
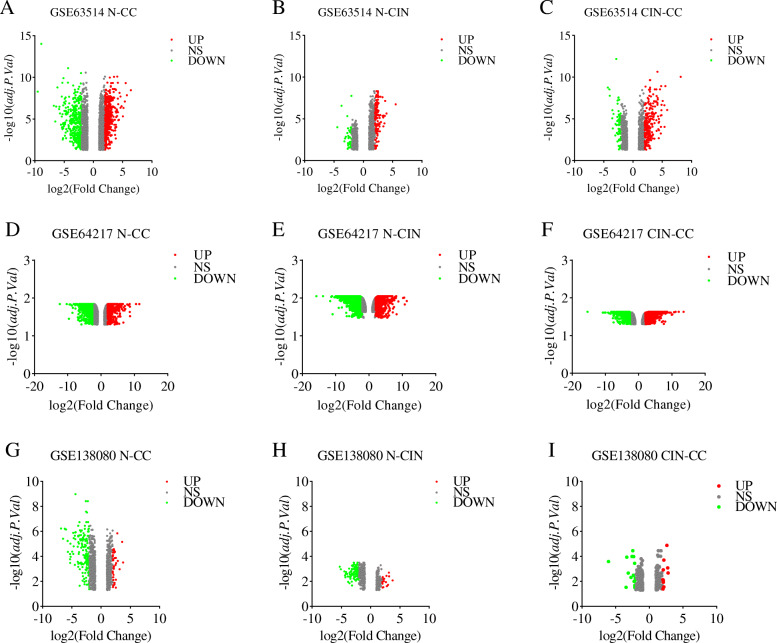
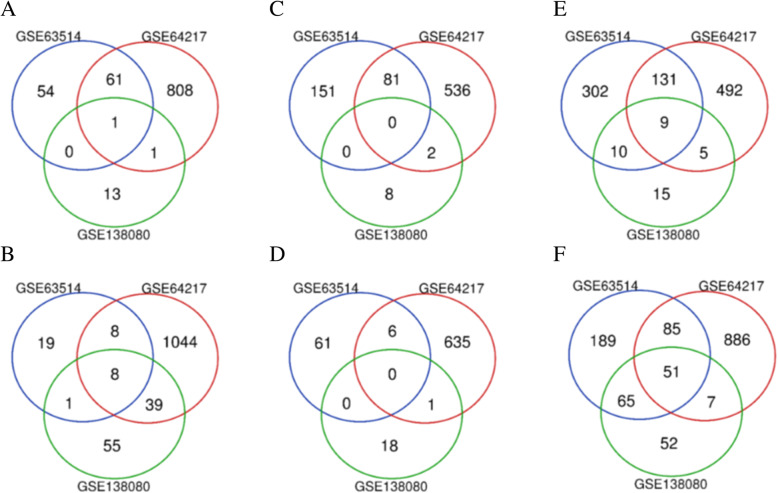
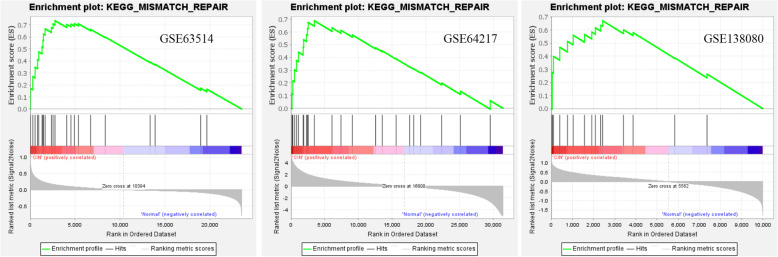
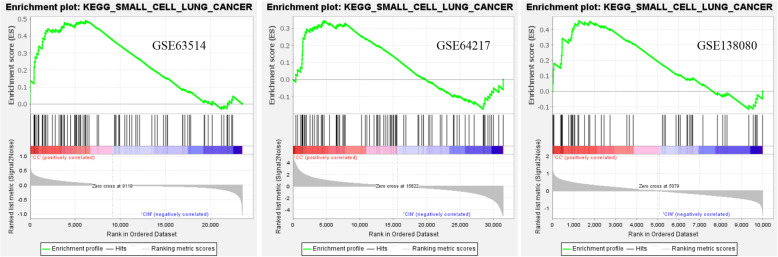
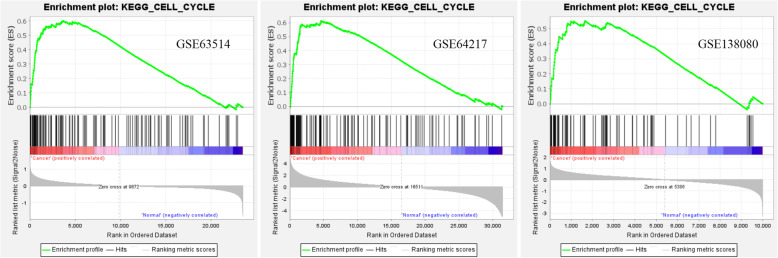
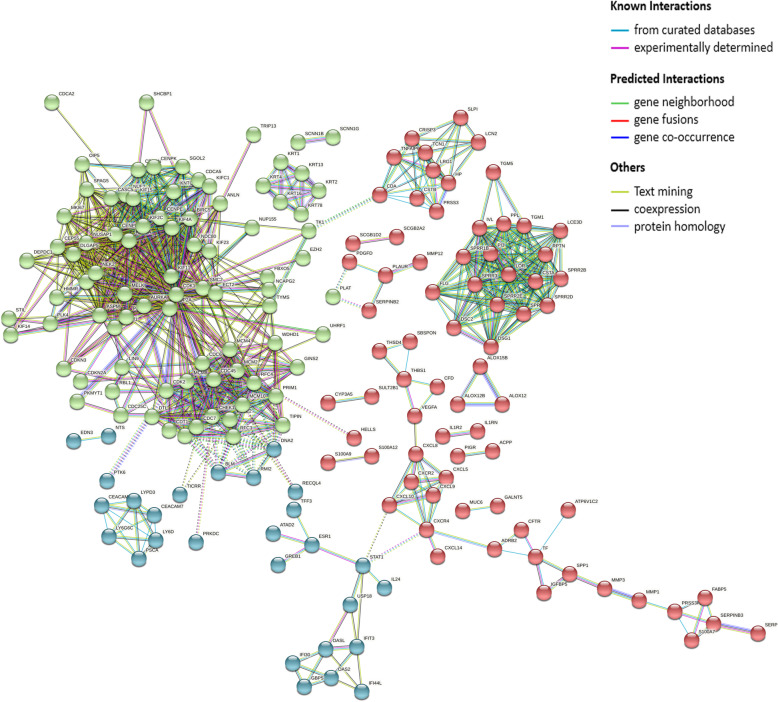
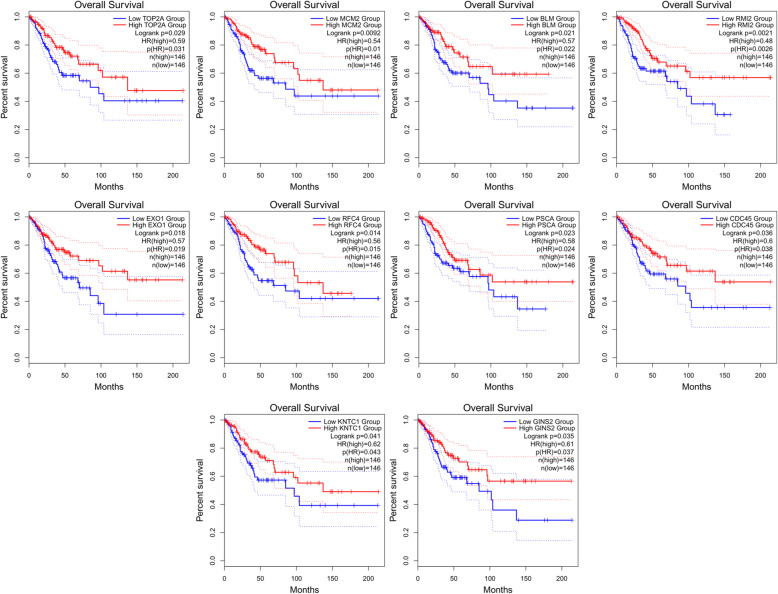
Three gene expression profiles (GSE63514, GSE64217 and GSE138080) were screened and downloaded from the Gene Expression Omnibus database (GEO). Differentially expressed genes (DEGs) were screened using the GEO2R and Venn diagram tools. Then, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed. Gene set enrichment analysis (GSEA) was performed to analyze the three gene expression profiles. Moreover, a protein-protein interaction (PPI) network of the DEGs was constructed, and functional enrichment analysis was performed. On this basis, hub genes from critical PPI subnetworks were explored with Cytoscape software. The expression of these genes in tumors was verified, and survival analysis of potential prognostic genes from critical subnetworks was conducted. Functional annotation, multiple gene comparison and dimensionality reduction in candidate genes indicated the clinical significance of potential targets.
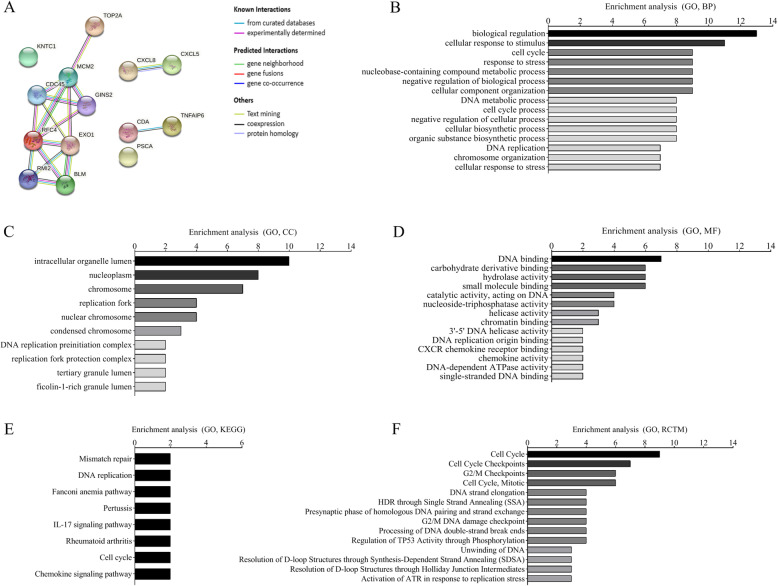
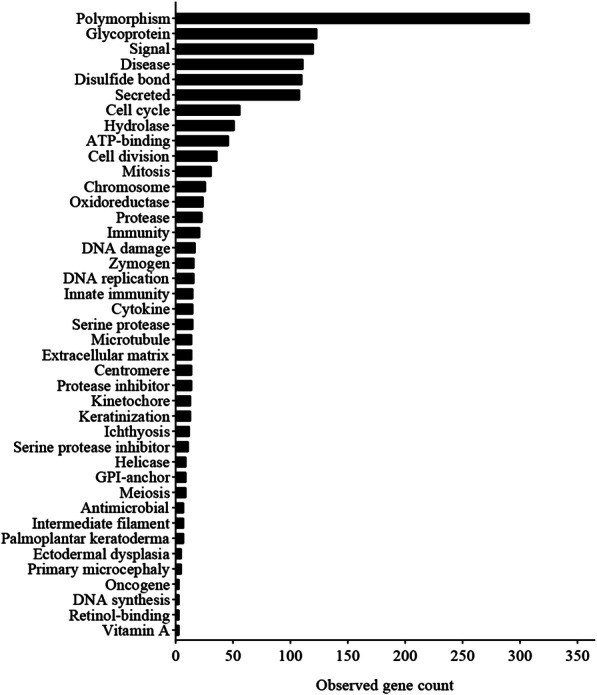
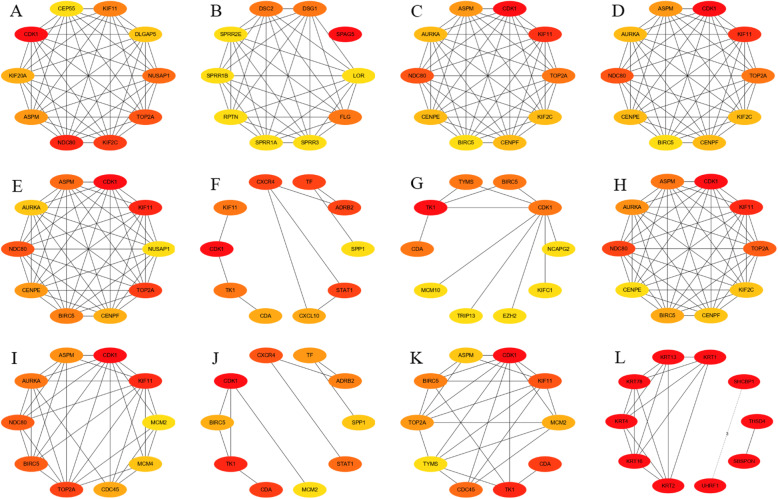
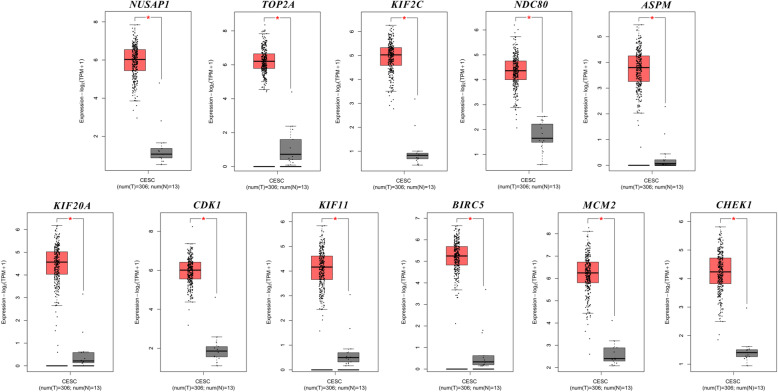
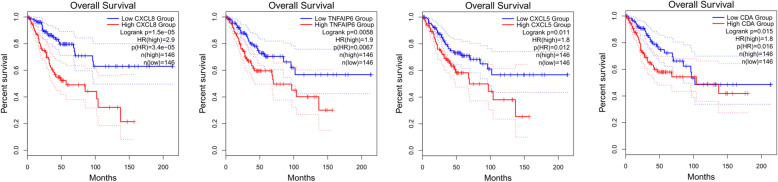
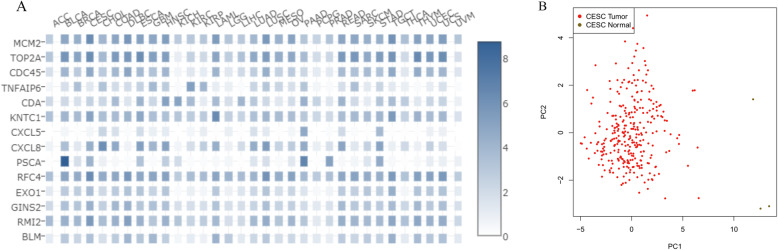
A total of 476 DEGs were screened: 253 upregulated genes and 223 downregulated genes. DEGs were enriched in 22 biological processes, 16 cellular components and 9 molecular functions in precancerous lesions and cervical cancer. DEGs were mainly enriched in 10 KEGG pathways. Through intersection analysis and data mining, 3 key KEGG pathways and related core genes were revealed by GSEA. Moreover, a PPI network of 476 DEGs was constructed, hub genes from 12 critical subnetworks were explored, and a total of 14 potential molecular targets were obtained.
These findings promote the understanding of the molecular mechanism of and clinically related molecular targets for cervical cancer.
本研究旨在探索和确定导致宫颈癌进展的关键基因和信号通路,以改善预后。
从基因表达综合数据库(GEO)中筛选并下载了三个基因表达谱(GSE63514、GSE64217 和 GSE138080)。使用 GEO2R 和 Venn 图工具筛选差异表达基因(DEGs)。然后进行基因本体论(GO)和京都基因与基因组百科全书(KEGG)通路富集分析。进行基因集富集分析(GSEA)以分析三个基因表达谱。此外,构建 DEGs 的蛋白质-蛋白质相互作用(PPI)网络,并进行功能富集分析。在此基础上,利用 Cytoscape 软件探索关键 PPI 子网络中的枢纽基因。验证这些基因在肿瘤中的表达,并对关键子网络中的潜在预后基因进行生存分析。对候选基因进行功能注释、多基因比较和降维分析,表明了潜在靶标的临床意义。
共筛选出 476 个 DEGs:253 个上调基因和 223 个下调基因。DEGs 在癌前病变和宫颈癌中富集于 22 个生物过程、16 个细胞成分和 9 个分子功能。DEGs 主要富集于 10 个 KEGG 通路。通过交集分析和数据挖掘,通过 GSEA 揭示了 3 个关键 KEGG 通路和相关的核心基因。此外,构建了 476 个 DEGs 的 PPI 网络,探索了 12 个关键子网的枢纽基因,并获得了 14 个潜在的分子靶标。
这些发现促进了对宫颈癌分子机制和临床相关分子靶标的理解。