Johns Hopkins Bloomberg School of Public Health, Department of Epidemiology, Baltimore, Maryland, United States of America.
PLoS Comput Biol. 2021 Jul 6;17(7):e1009182. doi: 10.1371/journal.pcbi.1009182. eCollection 2021 Jul.
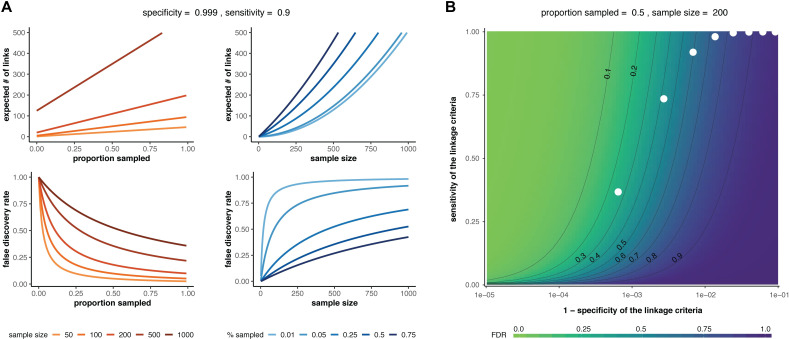
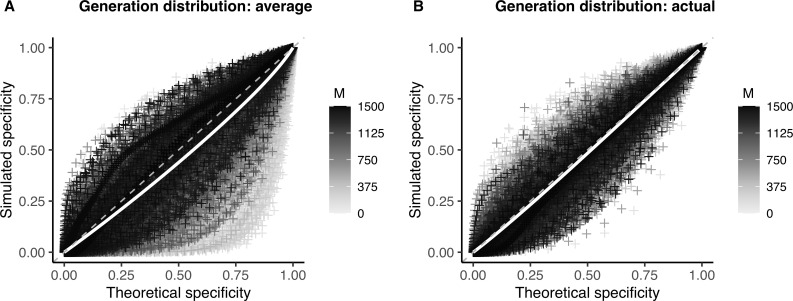
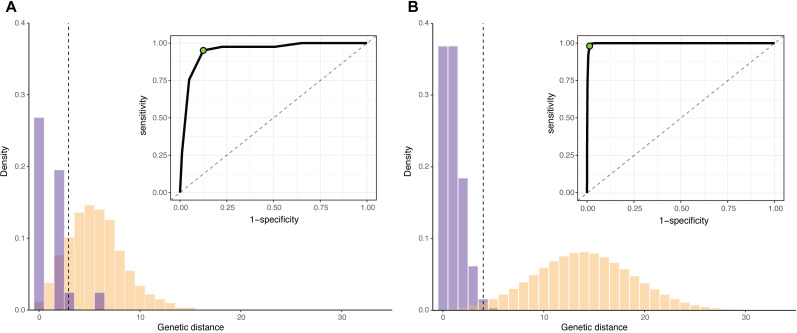
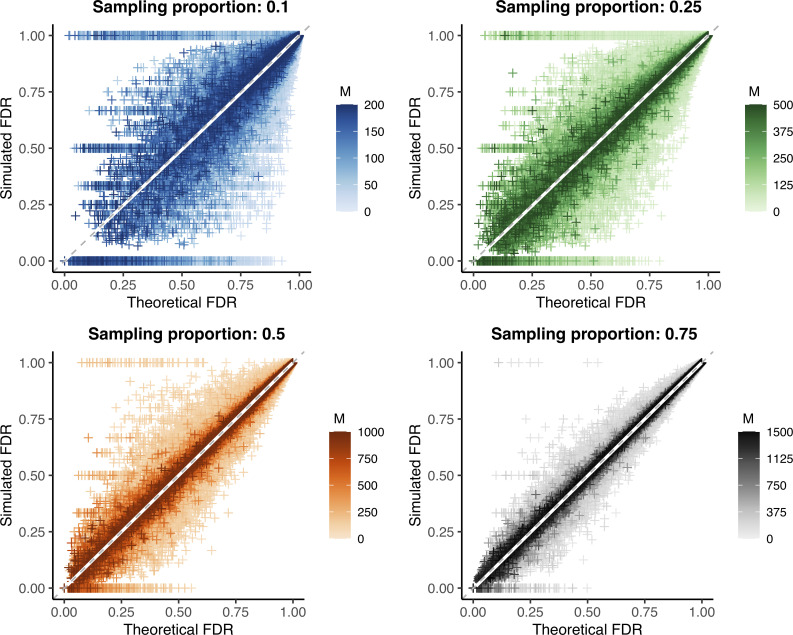
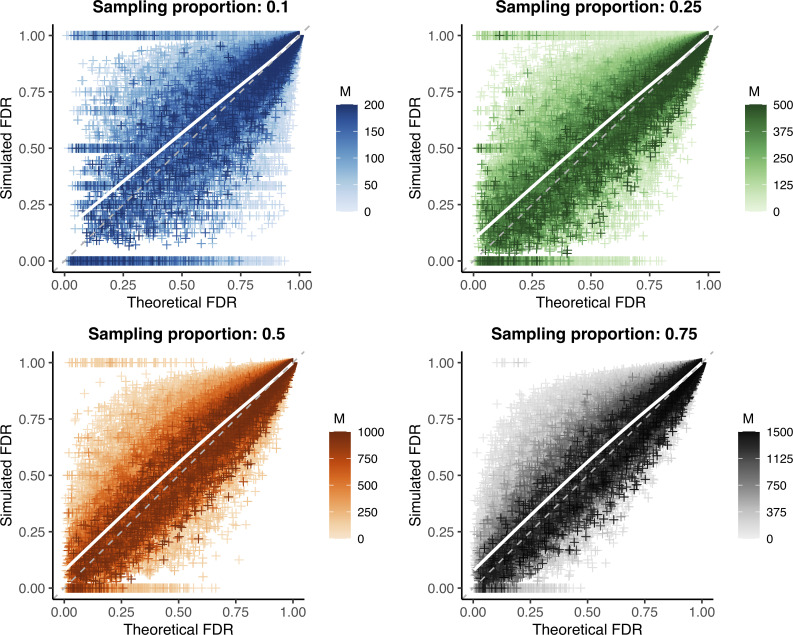
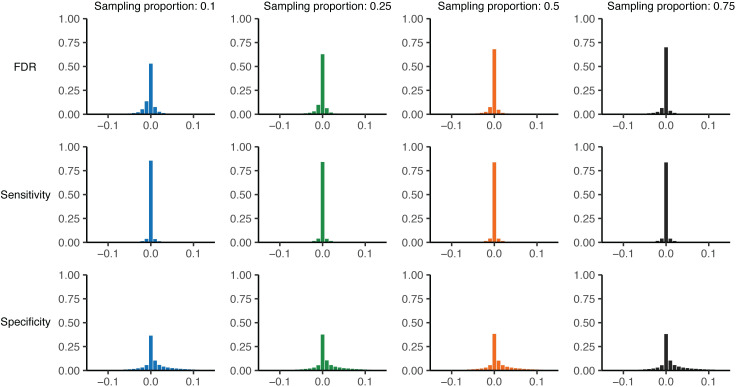
Sample size calculations are an essential component of the design and evaluation of scientific studies. However, there is a lack of clear guidance for determining the sample size needed for phylogenetic studies, which are becoming an essential part of studying pathogen transmission. We introduce a statistical framework for determining the number of true infector-infectee transmission pairs identified by a phylogenetic study, given the size and population coverage of that study. We then show how characteristics of the criteria used to determine linkage and aspects of the study design can influence our ability to correctly identify transmission links, in sometimes counterintuitive ways. We test the overall approach using outbreak simulations and provide guidance for calculating the sensitivity and specificity of the linkage criteria, the key inputs to our approach. The framework is freely available as the R package phylosamp, and is broadly applicable to designing and evaluating a wide array of pathogen phylogenetic studies.
样本量计算是科学研究设计和评估的重要组成部分。然而,对于确定系统发育研究所需的样本量,缺乏明确的指导,而系统发育研究正成为研究病原体传播的重要组成部分。我们引入了一个统计框架,用于确定给定研究的大小和人群覆盖范围的情况下,通过系统发育研究确定的真实感染者-感染者传播对的数量。然后,我们展示了用于确定关联的标准的特征以及研究设计的各个方面如何以有时违反直觉的方式影响我们正确识别传播联系的能力。我们使用暴发模拟测试了整体方法,并提供了用于计算关联标准的灵敏度和特异性的指导,这是我们方法的关键输入。该框架作为 R 包 phylosamp 免费提供,并且广泛适用于设计和评估各种病原体系统发育研究。