Rahmatbakhsh Matineh, Gagarinova Alla, Babu Mohan
Department of Biochemistry, University of Regina, Regina, SK, Canada.
Department of Biochemistry, Microbiology, & Immunology, University of Saskatchewan, Saskatoon, SK, Canada.
Front Genet. 2021 Jul 2;12:667936. doi: 10.3389/fgene.2021.667936. eCollection 2021.
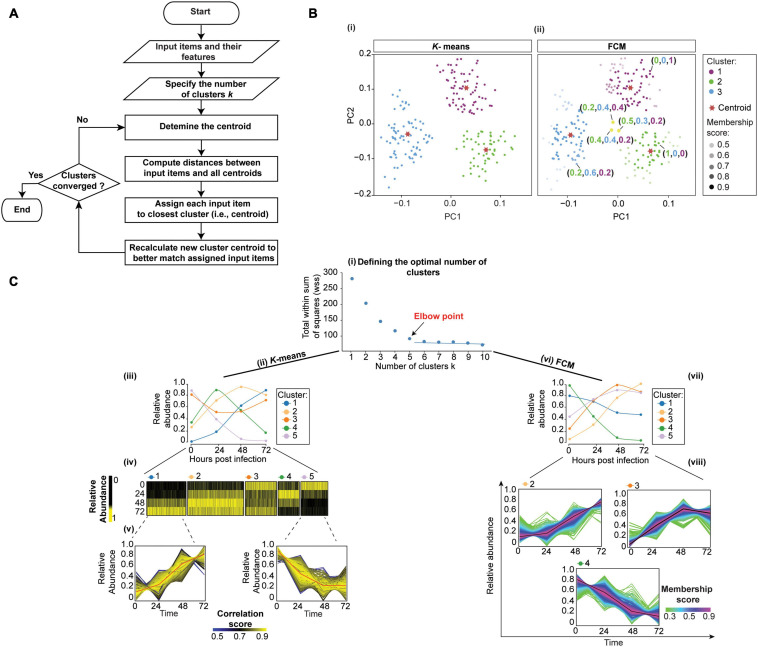
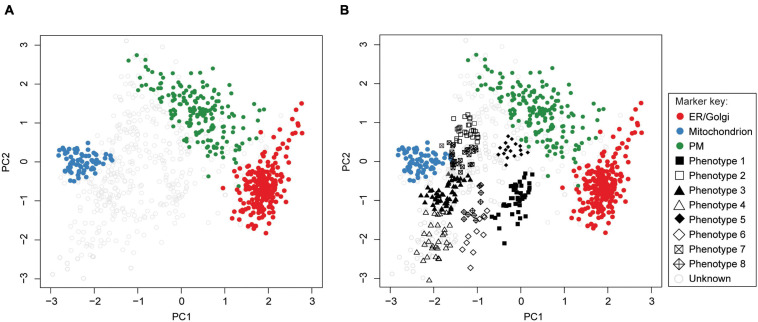
Microbial pathogens have evolved numerous mechanisms to hijack host's systems, thus causing disease. This is mediated by alterations in the combined host-pathogen proteome in time and space. Mass spectrometry-based proteomics approaches have been developed and tailored to map disease progression. The result is complex multidimensional data that pose numerous analytic challenges for downstream interpretation. However, a systematic review of approaches for the downstream analysis of such data has been lacking in the field. In this review, we detail the steps of a typical temporal and spatial analysis, including data pre-processing steps (i.e., quality control, data normalization, the imputation of missing values, and dimensionality reduction), different statistical and machine learning approaches, validation, interpretation, and the extraction of biological information from mass spectrometry data. We also discuss current best practices for these steps based on a collection of independent studies to guide users in selecting the most suitable strategies for their dataset and analysis objectives. Moreover, we also compiled the list of commonly used R software packages for each step of the analysis. These could be easily integrated into one's analysis pipeline. Furthermore, we guide readers through various analysis steps by applying these workflows to mock and host-pathogen interaction data from public datasets. The workflows presented in this review will serve as an introduction for data analysis novices, while also helping established users update their data analysis pipelines. We conclude the review by discussing future directions and developments in temporal and spatial proteomics and data analysis approaches. Data analysis codes, prepared for this review are available from https://github.com/BabuLab-UofR/TempSpac, where guidelines and sample datasets are also offered for testing purposes.
微生物病原体已经进化出多种机制来劫持宿主系统,从而引发疾病。这是通过宿主 - 病原体蛋白质组在时间和空间上的变化来介导的。基于质谱的蛋白质组学方法已经得到开发和定制,以描绘疾病进展情况。结果是复杂的多维数据,给下游解释带来了众多分析挑战。然而,该领域一直缺乏对此类数据下游分析方法的系统综述。在本综述中,我们详细介绍了典型的时空分析步骤,包括数据预处理步骤(即质量控制、数据归一化、缺失值插补和降维)、不同的统计和机器学习方法、验证、解释以及从质谱数据中提取生物信息。我们还基于一系列独立研究讨论了这些步骤的当前最佳实践,以指导用户为其数据集和分析目标选择最合适的策略。此外,我们还编制了分析每个步骤常用的R软件包列表。这些可以很容易地集成到个人的分析流程中。此外,我们通过将这些工作流程应用于来自公共数据集的模拟数据和宿主 - 病原体相互作用数据,引导读者完成各种分析步骤。本综述中介绍的工作流程将为数据分析新手提供入门指导,同时也帮助有经验的用户更新他们的数据分析流程。我们通过讨论时空蛋白质组学和数据分析方法的未来方向和发展来结束本综述。为本次综述准备的数据分析代码可从https://github.com/BabuLab-UofR/TempSpac获取,该网站还提供了用于测试目的的指南和示例数据集。