Oslo Center for Biostatistics and Epidemiology, UiO, University of Oslo, Oslo, Norway.
Department of Community Medicine, UiT, The Arctic University of Norway, Tromsö, Norway.
BMC Bioinformatics. 2021 Aug 5;22(1):395. doi: 10.1186/s12859-021-04296-0.
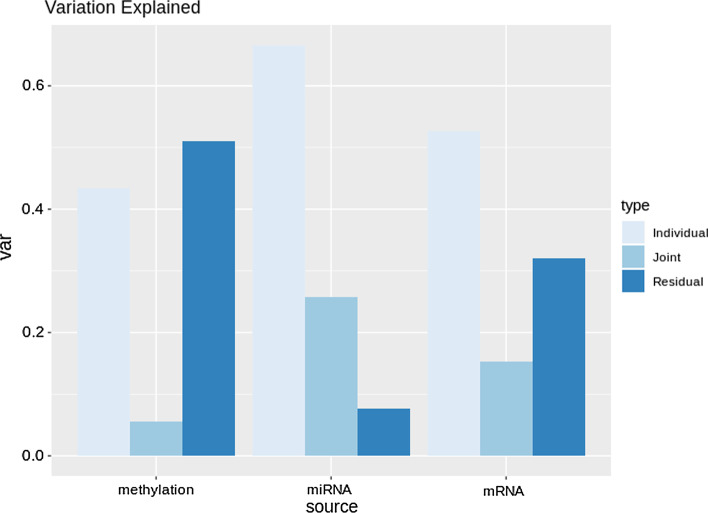
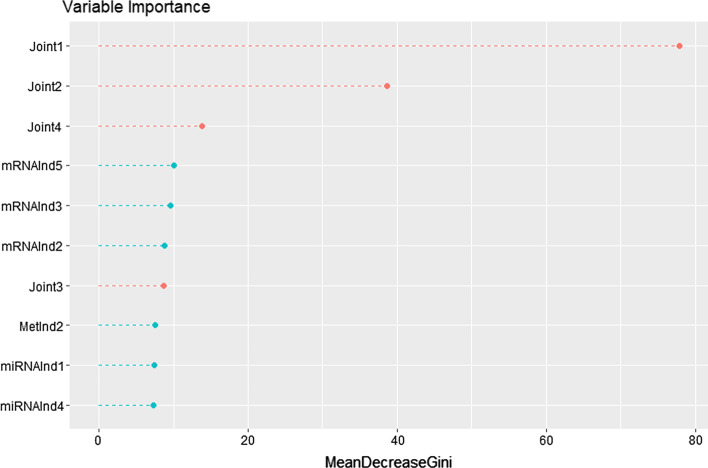
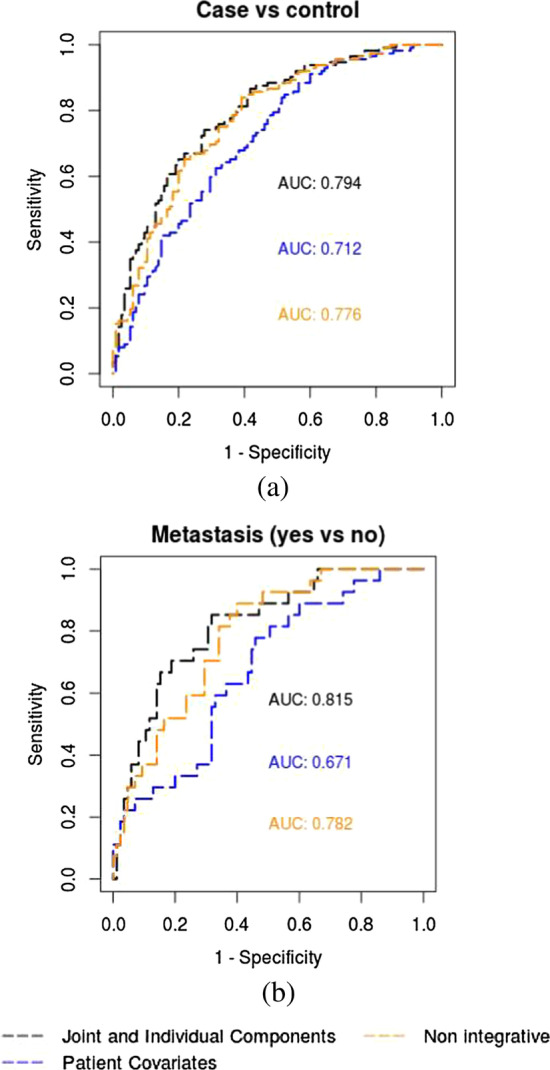
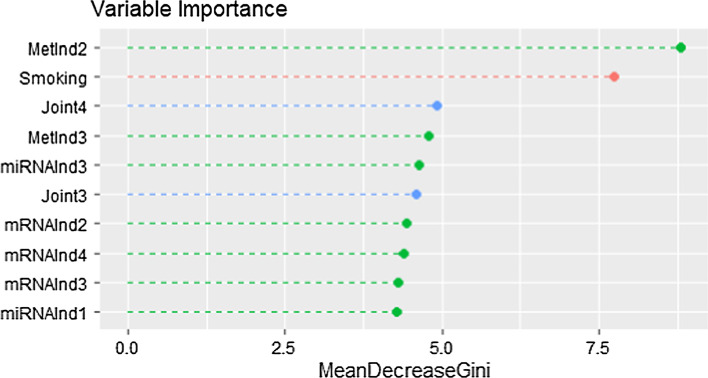
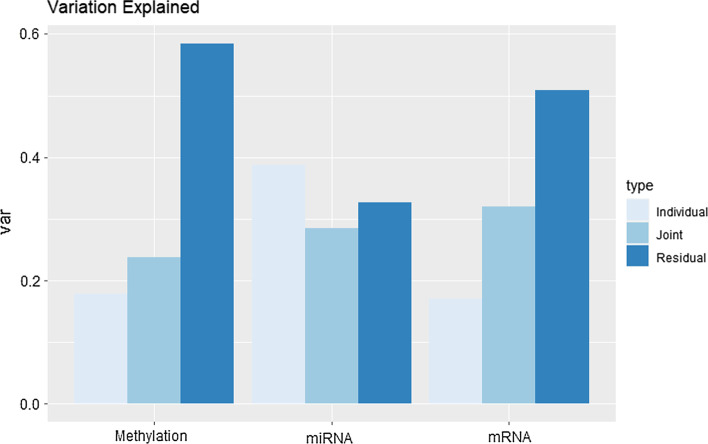
Cancer genomic studies often include data collected from several omics platforms. Each omics data source contributes to the understanding of the underlying biological process via source specific ("individual") patterns of variability. At the same time, statistical associations and potential interactions among the different data sources can reveal signals from common biological processes that might not be identified by single source analyses. These common patterns of variability are referred to as "shared" or "joint". In this work, we show how the use of joint and individual components can lead to better predictive models, and to a deeper understanding of the biological process at hand. We identify joint and individual contributions of DNA methylation, miRNA and mRNA expression collected from blood samples in a lung cancer case-control study nested within the Norwegian Women and Cancer (NOWAC) cohort study, and we use such components to build prediction models for case-control and metastatic status. To assess the quality of predictions, we compare models based on simultaneous, integrative analysis of multi-source omics data to a standard non-integrative analysis of each single omics dataset, and to penalized regression models. Additionally, we apply the proposed approach to a breast cancer dataset from The Cancer Genome Atlas.
Our results show how an integrative analysis that preserves both components of variation is more appropriate than standard multi-omics analyses that are not based on such a distinction. Both joint and individual components are shown to contribute to a better quality of model predictions, and facilitate the interpretation of the underlying biological processes in lung cancer development.
In the presence of multiple omics data sources, we recommend the use of data integration techniques that preserve the joint and individual components across the omics sources. We show how the inclusion of such components increases the quality of model predictions of clinical outcomes.
癌症基因组研究通常包括从多个组学平台收集的数据。每个组学数据源通过特定于源的(“个体”)可变性模式为理解潜在的生物学过程做出贡献。同时,不同数据源之间的统计关联和潜在相互作用可以揭示可能无法通过单源分析识别的常见生物学过程的信号。这些共同的可变性模式被称为“共享”或“联合”。在这项工作中,我们展示了如何使用联合和个体成分来构建更好的预测模型,并深入了解手头的生物学过程。我们确定了在嵌套于挪威妇女与癌症(NOWAC)队列研究中的肺癌病例对照研究中从血液样本中收集的 DNA 甲基化、miRNA 和 mRNA 表达的联合和个体贡献,并使用这些成分构建病例对照和转移状态的预测模型。为了评估预测的质量,我们将基于多组学数据的同时、综合分析的模型与每个单组学数据集的非综合分析以及惩罚回归模型进行比较。此外,我们将提出的方法应用于来自癌症基因组图谱的乳腺癌数据集。
我们的结果表明,与不基于这种区别的标准多组学分析相比,保留变异的两个成分的综合分析更合适。联合和个体成分都被证明有助于提高模型预测的质量,并促进对肺癌发展中潜在生物学过程的解释。
在存在多个组学数据源的情况下,我们建议使用保留组学源之间联合和个体成分的数据集成技术。我们展示了包含这些成分如何提高临床结果模型预测的质量。