Faculty of Technical Sciences, Department of Agroecology, Aarhus University, Slagelse, Denmark.
PLoS One. 2021 Oct 26;16(10):e0259171. doi: 10.1371/journal.pone.0259171. eCollection 2021.
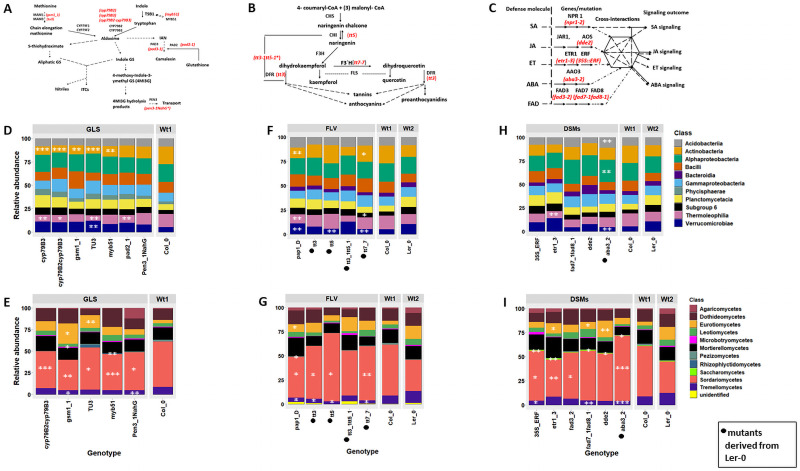
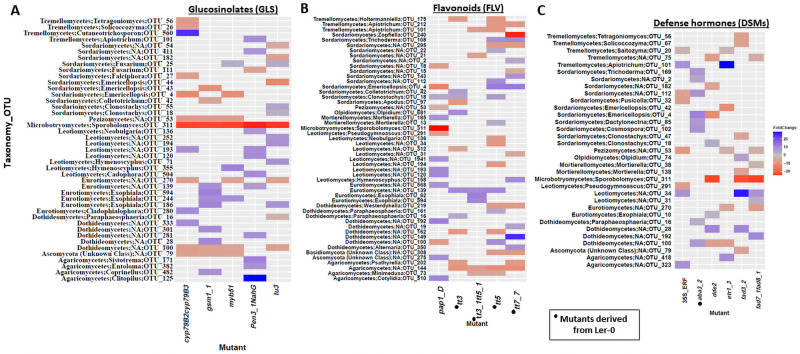
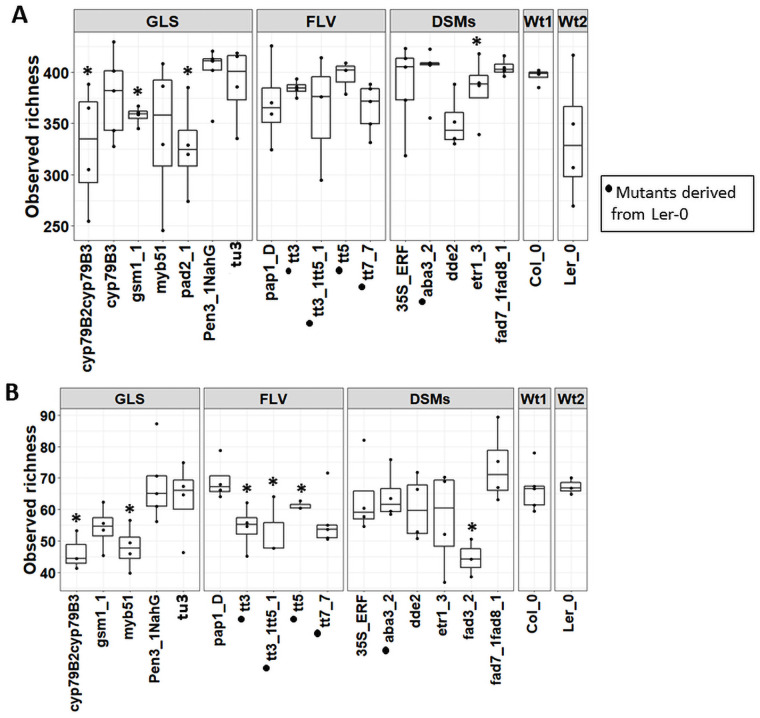
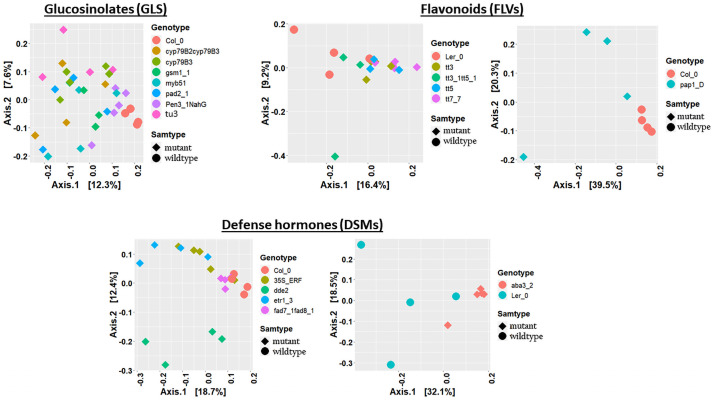
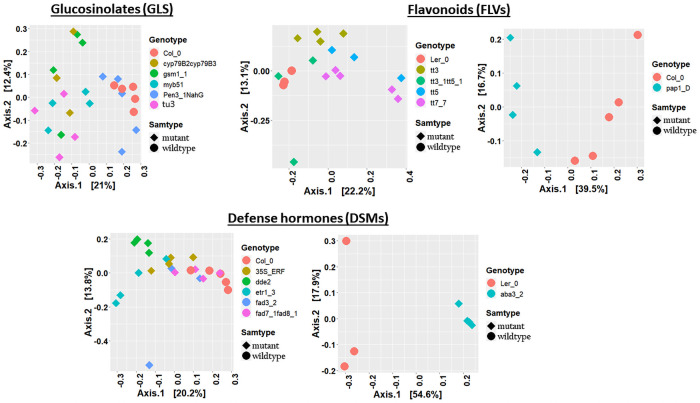
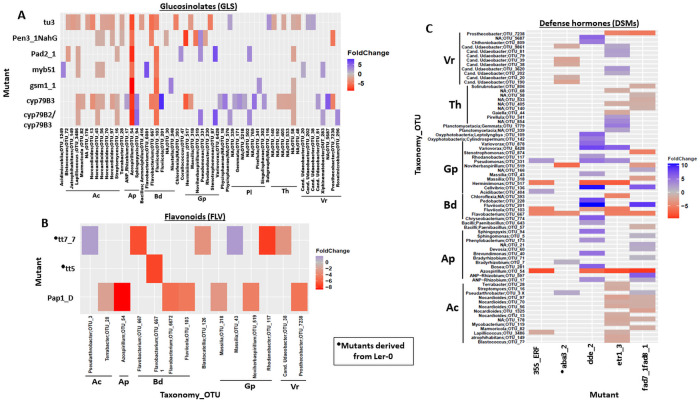
Plant associated microbiomes are known to confer fitness advantages to the host. Understanding how plant factors including biochemical traits influence host associated microbiome assembly could facilitate the development of microbiome-mediated solutions for sustainable plant production. Here, we examined microbial community structures of a set of well-characterized Arabidopsis thaliana mutants disrupted in metabolic pathways for the production of glucosinolates, flavonoids, or a number of defense signalling molecules. A. thaliana lines were grown in a natural soil and maintained under greenhouse conditions for 4 weeks before collection of roots for bacterial and fungal community profiling. We found distinct relative abundances and diversities of bacterial and fungal communities assembled in the individual A. thaliana mutants compared to their parental lines. Bacterial and fungal genera were mostly enriched than depleted in secondary metabolite and defense signaling mutants, except for flavonoid mutations on fungi communities. Bacterial genera Azospirillum and Flavobacterium were significantly enriched in most of the glucosinolate, flavonoid and signalling mutants while the fungal taxa Sporobolomyces and Emericellopsis were enriched in several glucosinolates and signalling mutants. Whilst the present study revealed marked differences in microbiomes of Arabidopsis mutants and their parental lines, it is suggestive that unknown enzymatic and pleiotropic activities of the mutated genes could contribute to the identified host-associated microbiomes. Notwithstanding, this study revealed interesting gene-microbiota links, and thus represents valuable resource data for selecting candidate A. thaliana mutants for analyzing the links between host genetics and the associated microbiome.
植物相关微生物组被认为赋予宿主适应优势。了解植物因素(包括生化特征)如何影响宿主相关微生物组的组装,可以促进开发微生物组介导的可持续植物生产解决方案。在这里,我们研究了一组代谢途径中生产硫代葡萄糖苷、类黄酮或多种防御信号分子的拟南芥突变体的微生物群落结构。在收集根进行细菌和真菌群落分析之前,将拟南芥系在天然土壤中生长并在温室条件下维持 4 周。与亲本系相比,我们发现单个拟南芥突变体中组装的细菌和真菌群落具有明显不同的相对丰度和多样性。除了真菌群落中的类黄酮突变外,次生代谢物和防御信号突变体中的细菌和真菌属主要是富集而不是耗尽。在大多数硫代葡萄糖苷、类黄酮和信号突变体中,细菌属 Azospirillum 和 Flavobacterium 显著富集,而真菌类群 Sporobolomyces 和 Emericellopsis 在几种硫代葡萄糖苷和信号突变体中富集。虽然本研究揭示了拟南芥突变体及其亲本系微生物组之间的明显差异,但暗示突变基因的未知酶和多效性活性可能有助于鉴定出的与宿主相关的微生物组。尽管如此,本研究揭示了有趣的基因-微生物群联系,因此代表了选择候选拟南芥突变体来分析宿主遗传学与相关微生物组之间联系的有价值的资源数据。