Department of Physiology, University of Arizona, Tucson, Arizona, United States of America.
Department of Medicine, Division of Endocrinology, University of Arizona, Tucson, Arizona, United States of America.
PLoS One. 2021 Oct 29;16(10):e0259449. doi: 10.1371/journal.pone.0259449. eCollection 2021.
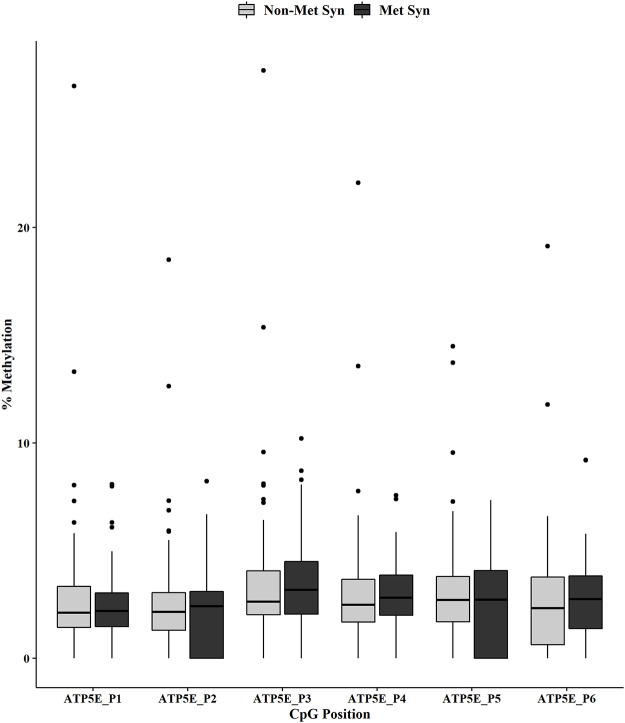
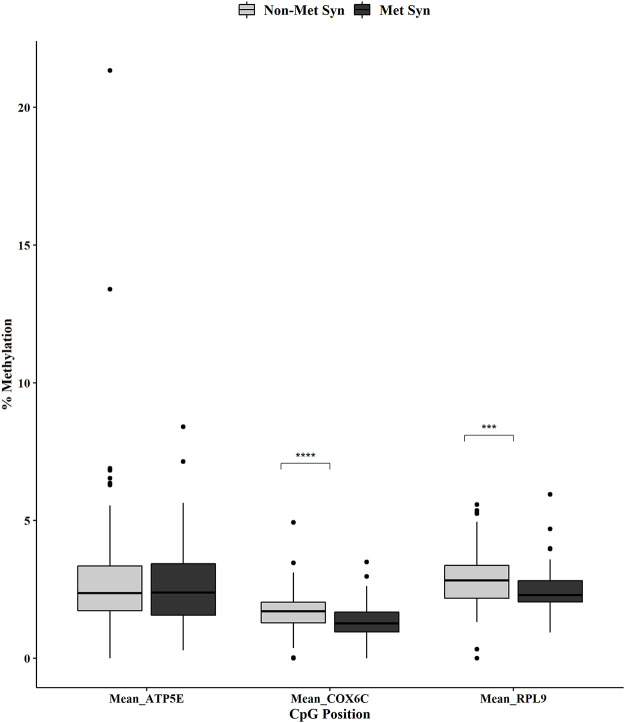
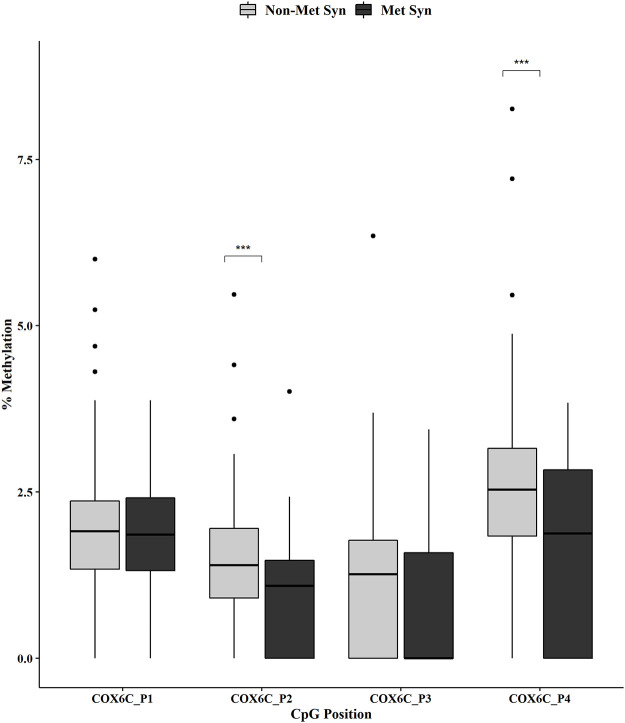
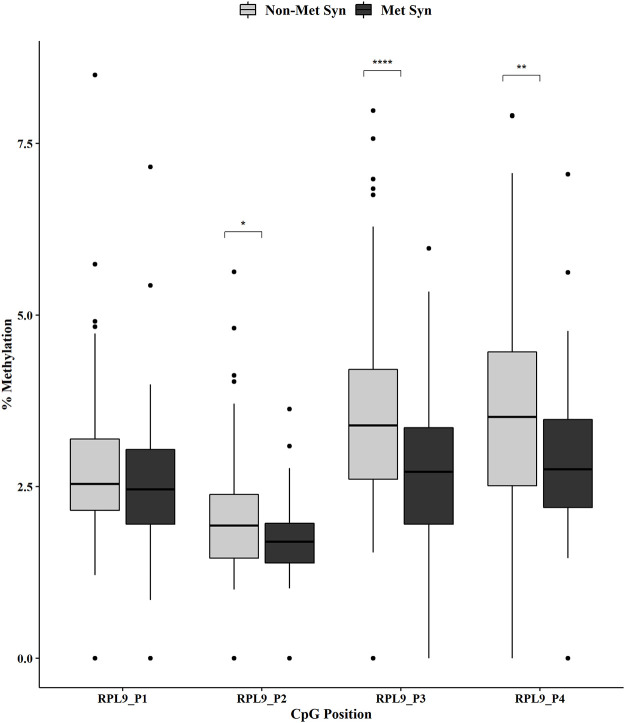
Metabolic syndrome (MetS) is highly prevalent worldwide. In the United States, estimates show that more than 30% of the adult population has MetS. MetS consists of multiple phenotypes, including obesity, dyslipidemia, and impaired glucose tolerance. Therefore, identifying the molecular mechanisms to explain this complex disease is critical for diagnosing and treating MetS. We previously showed 70 increased genes and 20 decreased genes in whole blood in MetS participants. The present study aimed to identify blood-based DNA methylation biomarkers in non-MetS versus MetS participants. The present study analyzed whole blood DNA samples from 184 adult participants of Latino descent from the Arizona Insulin Resistance (AIR) registry. We used the National Cholesterol Education Program Adult Treatment Panel III (NCEP: ATP III) criteria to identify non-MetS (n = 110) and MetS (n = 74) participants. We performed whole blood methylation analysis on select genes: ATP Synthase, H+ Transporting mitochondrial F1 Complex, Epsilon Subunit (ATP5E), Cytochrome C Oxidase Subunit VIc (COX6C), and Ribosomal Protein L9 (RPL9). The pyrosequencing analysis was a targeted approach focusing on the promoter region of each gene that specifically captured CpG methylation sites. In MetS participants, we showed decreased methylation in two CpG sites in COX6C and three CpG sites in RPL9, all p < 0.05 using the Mann-Whitney U test. There were no ATP5E CpG sites differently methylated in the MetS participants. Furthermore, while adjusting for age, gender, and smoking status, logistic regression analysis reaffirmed the associations between MetS and mean methylation within COX6C and RPL9 (both p < 0.05). In addition, Spearman's correlation revealed a significant inverse relationship between the previously published gene expression data and methylation data for RPL9 (p < 0.05). In summary, these results highlight potential blood DNA methylation biomarkers for the MetS phenotype. However, future validation studies are warranted to strengthen our findings.
代谢综合征(MetS)在全球范围内广泛流行。在美国,据估计有超过 30%的成年人患有 MetS。MetS 由多种表型组成,包括肥胖、血脂异常和葡萄糖耐量受损。因此,确定解释这种复杂疾病的分子机制对于诊断和治疗 MetS 至关重要。我们之前在 MetS 参与者的全血中发现了 70 个上调基因和 20 个下调基因。本研究旨在鉴定非 MetS 与 MetS 参与者之间的血液 DNA 甲基化生物标志物。本研究分析了来自亚利桑那胰岛素抵抗 (AIR) 登记处的 184 名拉丁裔成年参与者的全血 DNA 样本。我们使用国家胆固醇教育计划成人治疗小组 III (NCEP: ATP III) 标准来识别非 MetS(n=110)和 MetS(n=74)参与者。我们对选定的基因进行了全血甲基化分析:ATP 合酶、H+转运线粒体 F1 复合物、Epsilon 亚基(ATP5E)、细胞色素 C 氧化酶亚基 VIc(COX6C)和核糖体蛋白 L9(RPL9)。焦磷酸测序分析是一种靶向方法,主要针对每个基因的启动子区域,专门捕获 CpG 甲基化位点。在 MetS 参与者中,我们发现 COX6C 中有两个 CpG 位点和 RPL9 中有三个 CpG 位点的甲基化程度降低,使用 Mann-Whitney U 检验,所有结果均 p<0.05。MetS 参与者中没有 ATP5E CpG 位点的甲基化程度不同。此外,在调整年龄、性别和吸烟状况后,逻辑回归分析再次证实了 MetS 与 COX6C 和 RPL9 内平均甲基化之间的关联(均 p<0.05)。此外,Spearman 相关性分析显示 RPL9 的基因表达数据与甲基化数据之间存在显著的负相关关系(p<0.05)。总之,这些结果突出了 MetS 表型的潜在血液 DNA 甲基化生物标志物。然而,需要进一步的验证研究来加强我们的发现。