Department of Biochemistry and Molecular Biology, Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria 3010, Australia.
Department of Infectious Diseases, Central Clinical School, Monash University, Melbourne, Victoria 3004, Australia.
Microb Genom. 2019 Jul;5(7). doi: 10.1099/mgen.0.000269. Epub 2019 May 20.
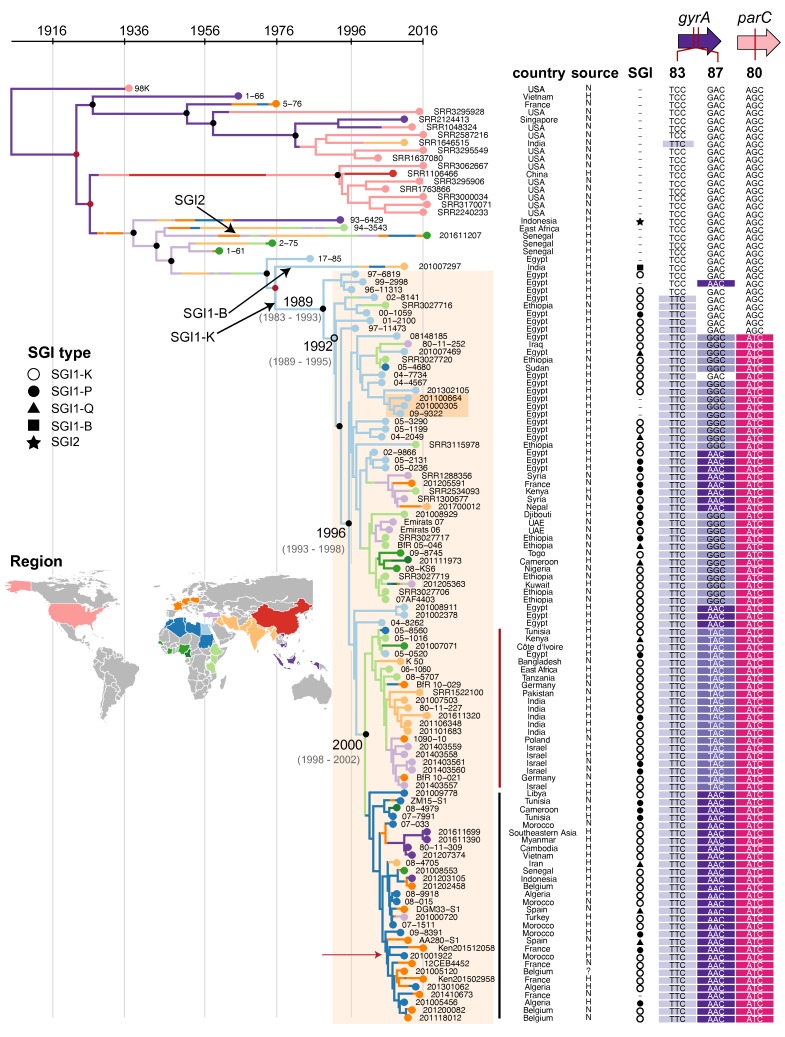
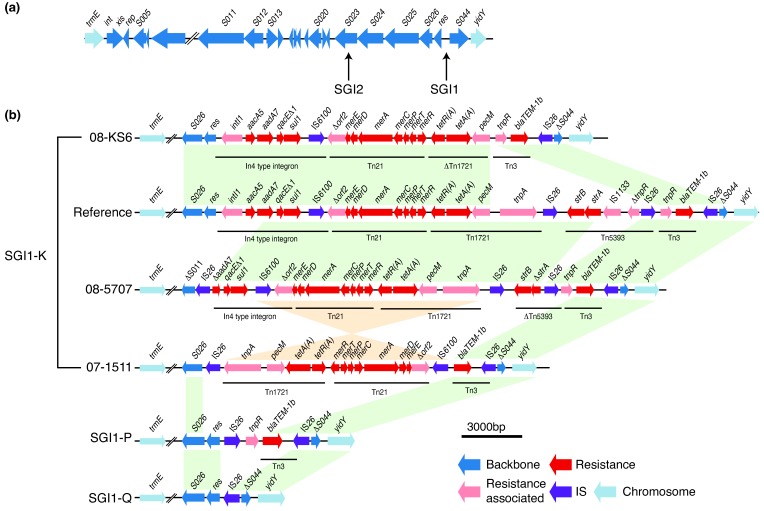
Salmonella enterica serotype Kentucky can be a common causative agent of salmonellosis, usually associated with consumption of contaminated poultry. Antimicrobial resistance (AMR) to multiple drugs, including ciprofloxacin, is an emerging problem within this serotype. We used whole-genome sequencing (WGS) to investigate the phylogenetic structure and AMR content of 121 S.enterica serotype Kentucky sequence type 198 isolates from five continents. Population structure was inferred using phylogenomic analysis and whole genomes were compared to investigate changes in gene content, with a focus on acquired AMR genes. Our analysis showed that multidrug-resistant (MDR) S.enterica serotype Kentucky isolates belonged to a single lineage, which we estimate emerged circa 1989 following the acquisition of the AMR-associated Salmonella genomic island (SGI) 1 (variant SGI1-K) conferring resistance to ampicillin, streptomycin, gentamicin, sulfamethoxazole and tetracycline. Phylogeographical analysis indicates this clone emerged in Egypt before disseminating into Northern, Southern and Western Africa, then to the Middle East, Asia and the European Union. The MDR clone has since accumulated various substitution mutations in the quinolone-resistance-determining regions (QRDRs) of DNA gyrase (gyrA) and DNA topoisomerase IV (parC), such that most strains carry three QRDR mutations which together confer resistance to ciprofloxacin. The majority of AMR genes in the S. enterica serotype Kentucky genomes were carried either on plasmids or SGI structures. Remarkably, each genome of the MDR clone carried a different SGI1-K derivative structure; this variation could be attributed to IS26-mediated insertions and deletions, which appear to have hampered previous attempts to trace the clone's evolution using sub-WGS resolution approaches. Several different AMR plasmids were also identified, encoding resistance to chloramphenicol, third-generation cephalosporins, carbapenems and/or azithromycin. These results indicate that most MDR S. enterica serotype Kentucky circulating globally result from the clonal expansion of a single lineage that acquired chromosomal AMR genes 30 years ago, and has continued to diversify and accumulate additional resistances to last-line oral antimicrobials. This article contains data hosted by Microreact.
肠炎沙门氏菌血清型肯塔基州通常是沙门氏菌病的常见病原体,通常与食用受污染的家禽有关。该血清型对包括环丙沙星在内的多种药物的耐药性(AMR)是一个新出现的问题。我们使用全基因组测序(WGS)来研究来自五大洲的 121 株肠炎沙门氏菌血清型 198 序列型的系统发育结构和 AMR 含量。通过基因组分析推断种群结构,并比较全基因组以研究基因内容的变化,重点是获得的 AMR 基因。我们的分析表明,多药耐药(MDR)肠炎沙门氏菌血清型肯塔基州分离株属于单一谱系,我们估计该谱系于 1989 年左右出现,当时获得了与 AMR 相关的沙门氏菌基因组岛(SGI)1(变体 SGI1-K),该岛赋予了对氨苄西林、链霉素、庆大霉素、磺胺甲恶唑和四环素的耐药性。系统发育地理分析表明,该克隆首先在埃及出现,然后传播到北非、南非和西非,然后传播到中东、亚洲和欧盟。自那时以来,MDR 克隆在喹诺酮耐药决定区(QRDR)的 DNA 回旋酶(gyrA)和 DNA 拓扑异构酶 IV(parC)中积累了各种取代突变,以至于大多数菌株携带三个 QRDR 突变,共同赋予对环丙沙星的耐药性。肠炎沙门氏菌血清型肯塔基州基因组中的大多数 AMR 基因要么位于质粒上,要么位于 SGI 结构上。值得注意的是,MDR 克隆的每个基因组都携带不同的 SGI1-K 衍生结构;这种变异可能归因于 IS26 介导的插入和缺失,这似乎阻碍了使用亚 WGS 分辨率方法追踪该克隆进化的先前尝试。还鉴定了几种不同的 AMR 质粒,编码对氯霉素、第三代头孢菌素、碳青霉烯类和/或阿奇霉素的耐药性。这些结果表明,全球循环的大多数 MDR 肠炎沙门氏菌血清型肯塔基州是由单一谱系的克隆扩张引起的,该谱系 30 年前获得了染色体 AMR 基因,并继续多样化和积累对最后一线口服抗菌药物的额外耐药性。本文包含由 Microreact 托管的数据。