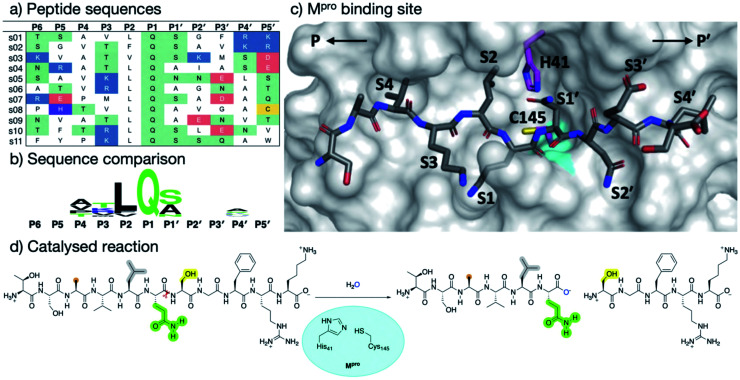
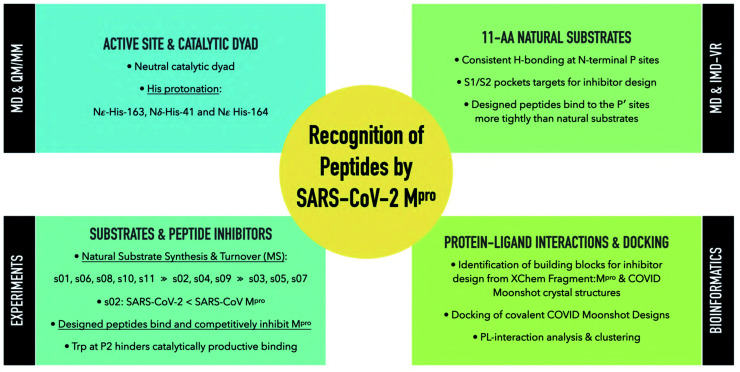
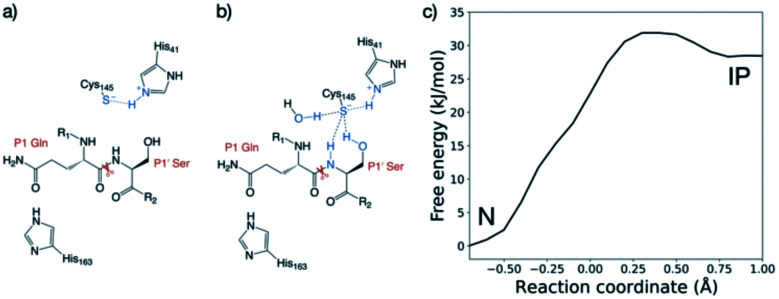
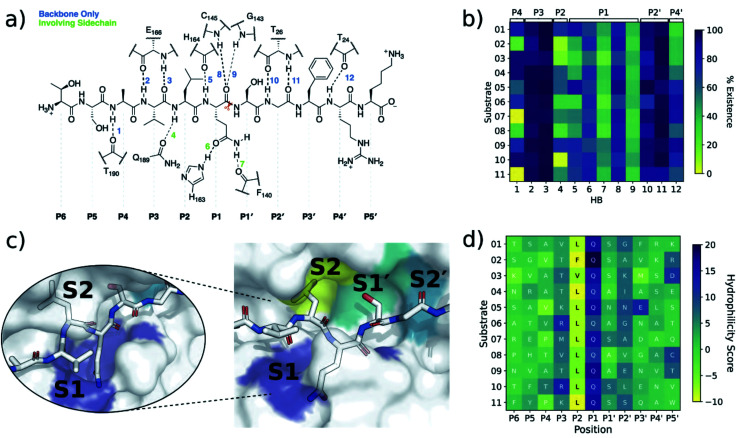
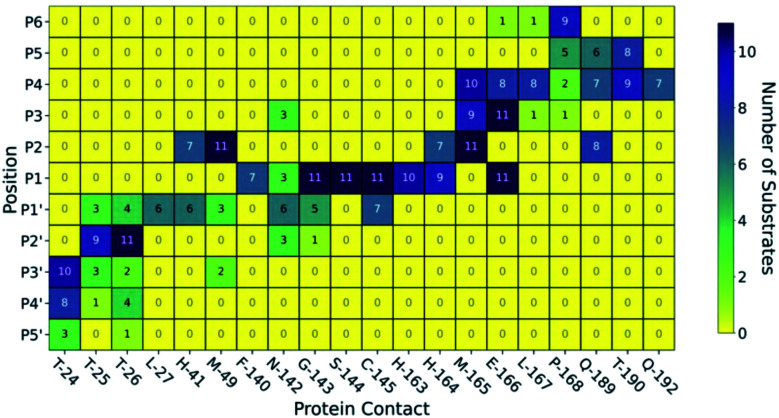
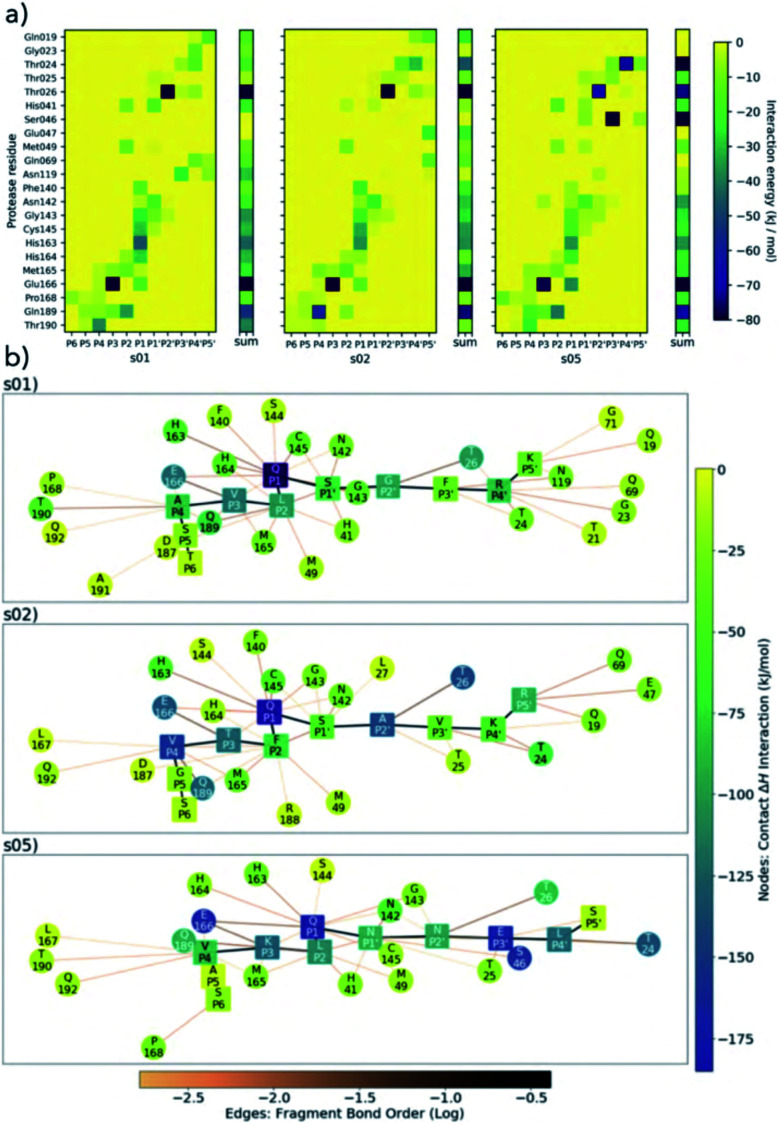
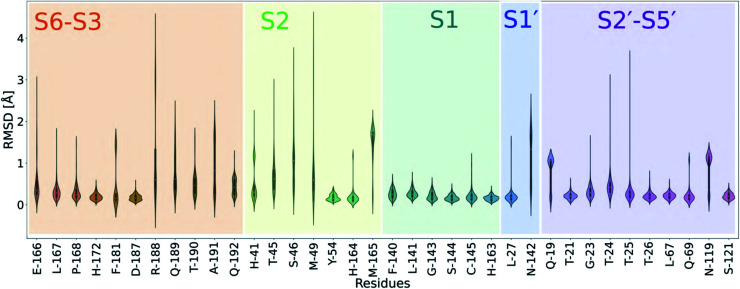
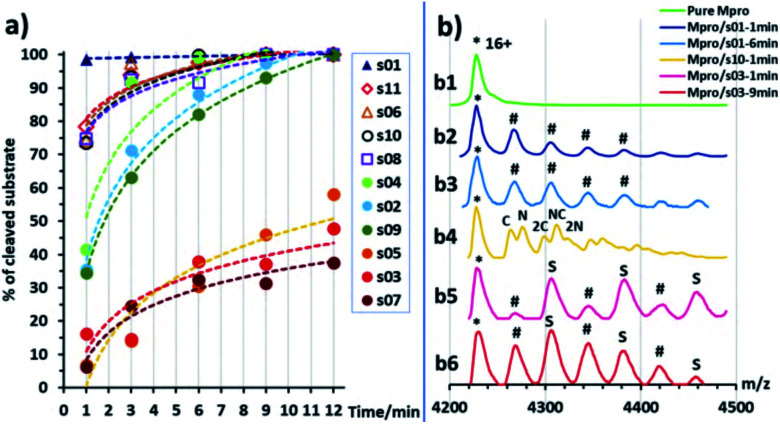
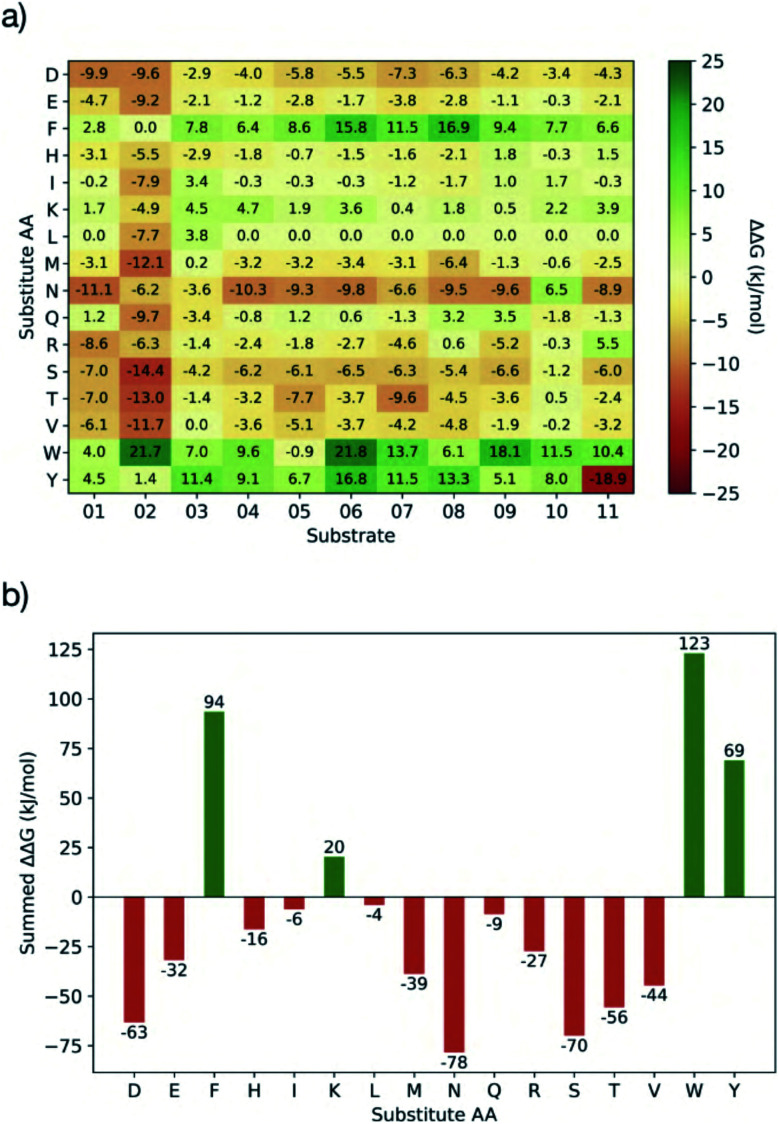
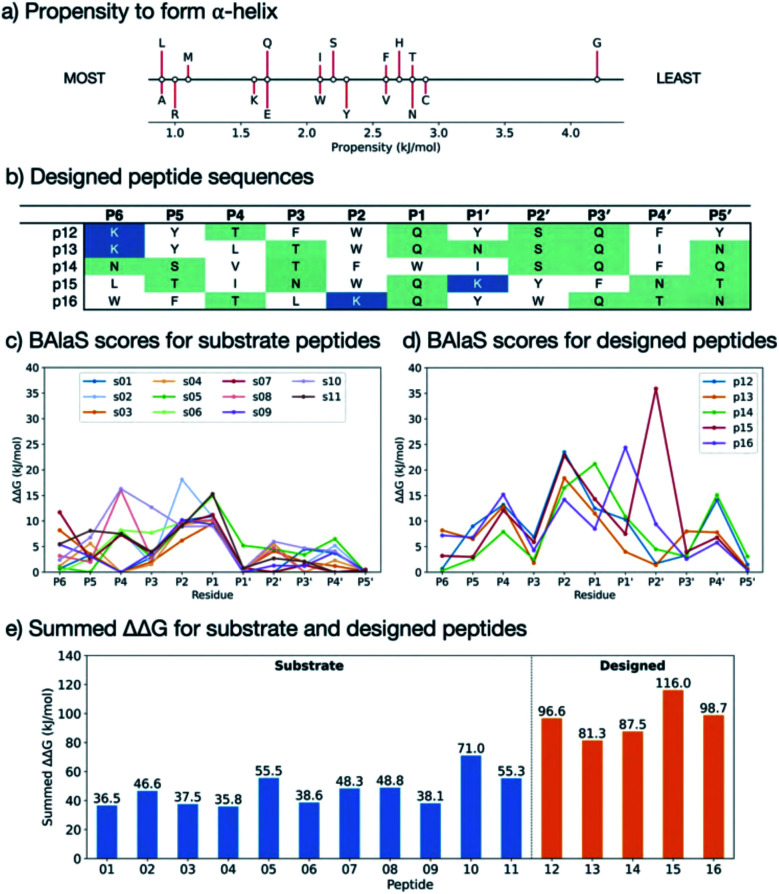
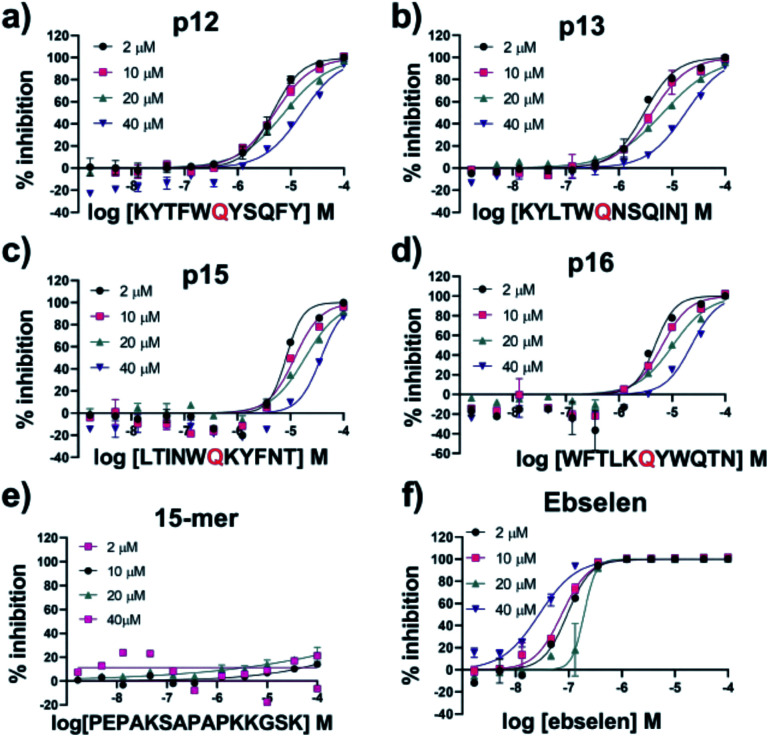
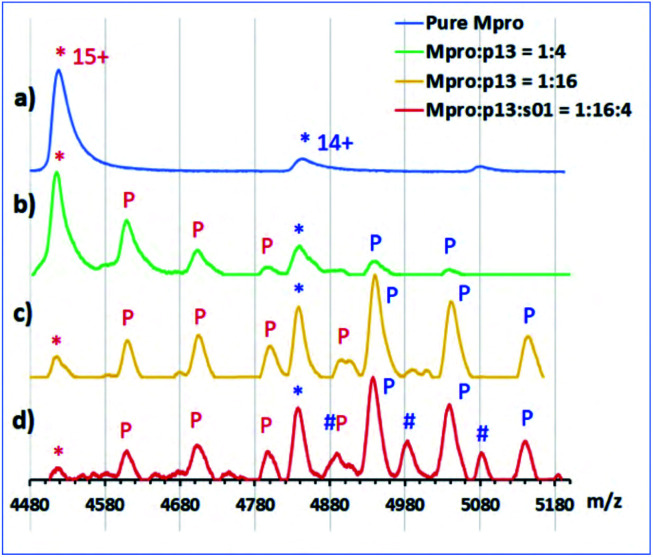
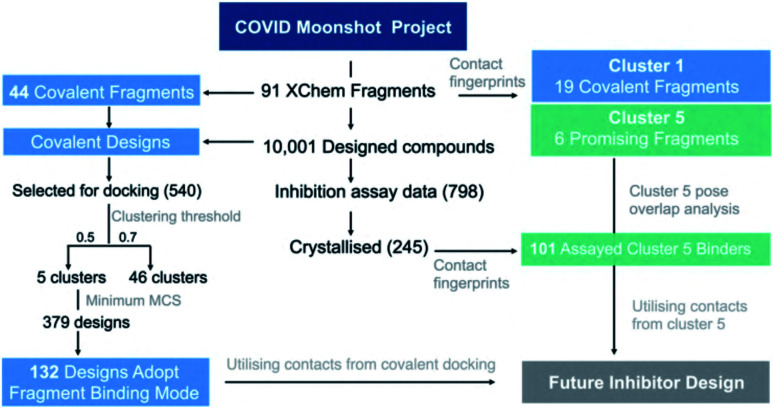
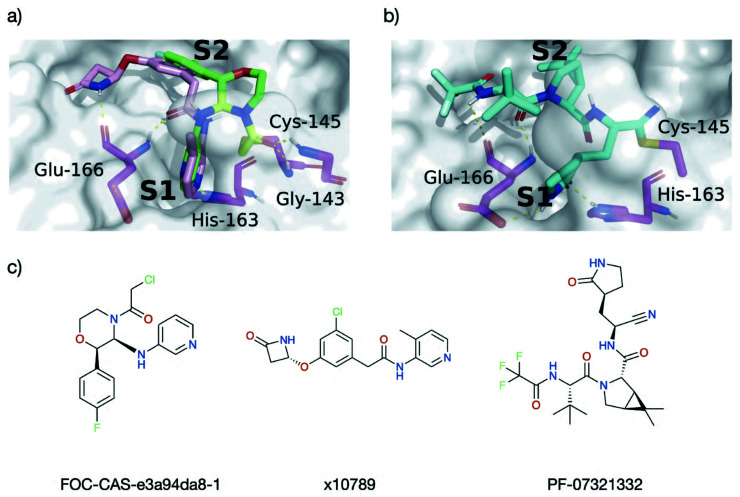
Chan H T Henry, Moesser Marc A, Walters Rebecca K, Malla Tika R, Twidale Rebecca M, John Tobias, Deeks Helen M, Johnston-Wood Tristan, Mikhailov Victor, Sessions Richard B, Dawson William, Salah Eidarus, Lukacik Petra, Strain-Damerell Claire, Owen C David, Nakajima Takahito, Świderek Katarzyna, Lodola Alessio, Moliner Vicent, Glowacki David R, Spencer James, Walsh Martin A, Schofield Christopher J, Genovese Luigi, Shoemark Deborah K, Mulholland Adrian J, Duarte Fernanda, Morris Garrett M
Chemistry Research Laboratory, Department of Chemistry and the Ineos Oxford Institute for Antimicrobial Research 12 Mansfield Road Oxford OX1 3TA UK
Department of Statistics, University of Oxford 24-29 St Giles' Oxford OX1 3LB UK
Chem Sci. 2021 Sep 6;12(41):13686-13703. doi: 10.1039/d1sc03628a. eCollection 2021 Oct 27.
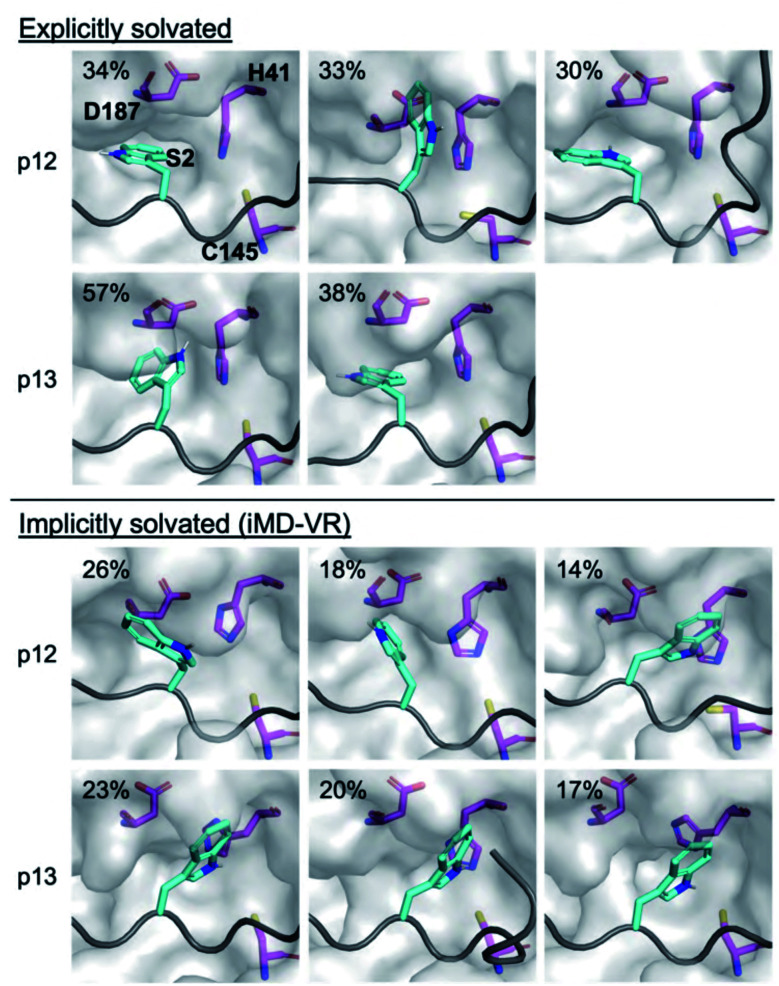
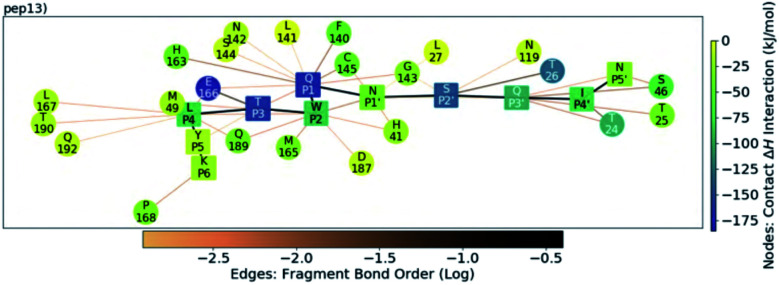
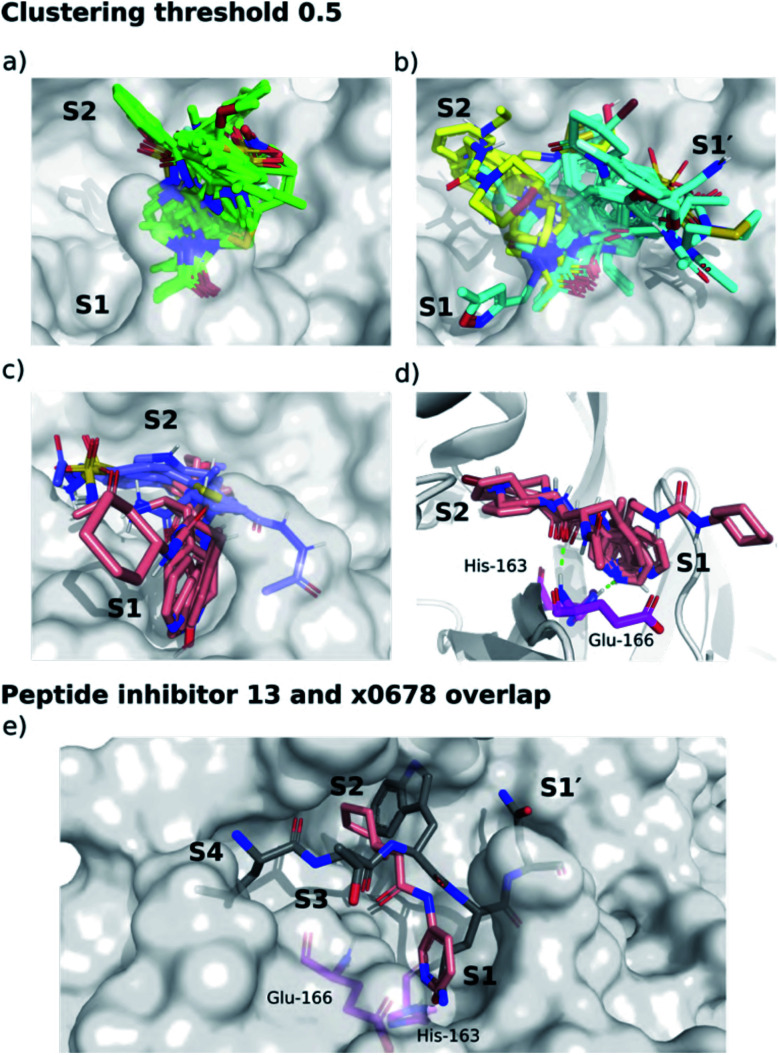
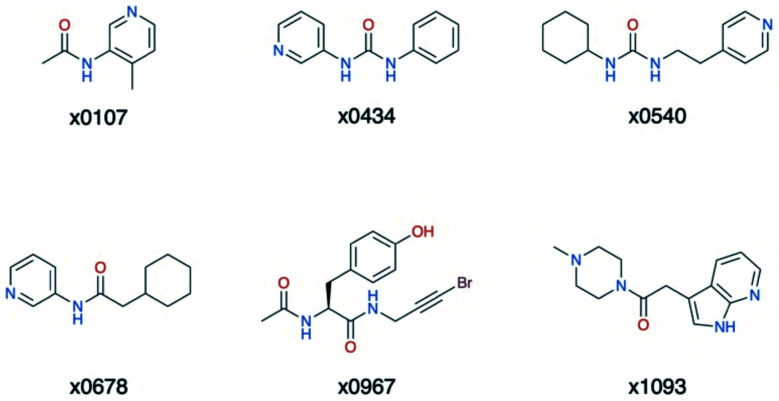
The main protease (M) of SARS-CoV-2 is central to viral maturation and is a promising drug target, but little is known about structural aspects of how it binds to its 11 natural cleavage sites. We used biophysical and crystallographic data and an array of biomolecular simulation techniques, including automated docking, molecular dynamics (MD) and interactive MD in virtual reality, QM/MM, and linear-scaling DFT, to investigate the molecular features underlying recognition of the natural M substrates. We extensively analysed the subsite interactions of modelled 11-residue cleavage site peptides, crystallographic ligands, and docked COVID Moonshot-designed covalent inhibitors. Our modelling studies reveal remarkable consistency in the hydrogen bonding patterns of the natural M substrates, particularly on the N-terminal side of the scissile bond. They highlight the critical role of interactions beyond the immediate active site in recognition and catalysis, in particular plasticity at the S2 site. Building on our initial M-substrate models, we used predictive saturation variation scanning (PreSaVS) to design peptides with improved affinity. Non-denaturing mass spectrometry and other biophysical analyses confirm these new and effective 'peptibitors' inhibit M competitively. Our combined results provide new insights and highlight opportunities for the development of M inhibitors as anti-COVID-19 drugs.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)的主要蛋白酶(M)对于病毒成熟至关重要,是一个很有前景的药物靶点,但对于它如何与其11个天然切割位点结合的结构方面却知之甚少。我们利用生物物理和晶体学数据以及一系列生物分子模拟技术,包括自动对接、分子动力学(MD)和虚拟现实中的交互式MD、量子力学/分子力学(QM/MM)以及线性标度密度泛函理论(DFT),来研究天然M底物识别背后的分子特征。我们广泛分析了模拟的11个残基切割位点肽、晶体学配体和对接的COVID Moonshot设计的共价抑制剂的亚位点相互作用。我们的建模研究揭示了天然M底物氢键模式的显著一致性,特别是在切割键的N端一侧。它们突出了直接活性位点之外的相互作用在识别和催化中的关键作用,特别是S2位点的可塑性。基于我们最初的M底物模型,我们使用预测性饱和变异扫描(PreSaVS)来设计具有更高亲和力的肽。非变性质谱和其他生物物理分析证实,这些新型有效的“肽抑制剂”竞争性地抑制M。我们的综合结果提供了新的见解,并突出了开发M抑制剂作为抗COVID-19药物的机会。