Pavlova Anna, Lynch Diane L, Daidone Isabella, Zanetti-Polzi Laura, Smith Micholas Dean, Chipot Chris, Kneller Daniel W, Kovalevsky Andrey, Coates Leighton, Golosov Andrei A, Dickson Callum J, Velez-Vega Camilo, Duca José S, Vermaas Josh V, Pang Yui Tik, Acharya Atanu, Parks Jerry M, Smith Jeremy C, Gumbart James C
School of Physics, Georgia Institute of Technology Atlanta GA 30332 USA
Department of Physical and Chemical Sciences, University of L'Aquila I-67010 L'Aquila Italy.
Chem Sci. 2020 Nov 26;12(4):1513-1527. doi: 10.1039/d0sc04942e. eCollection 2021 Jan 28.
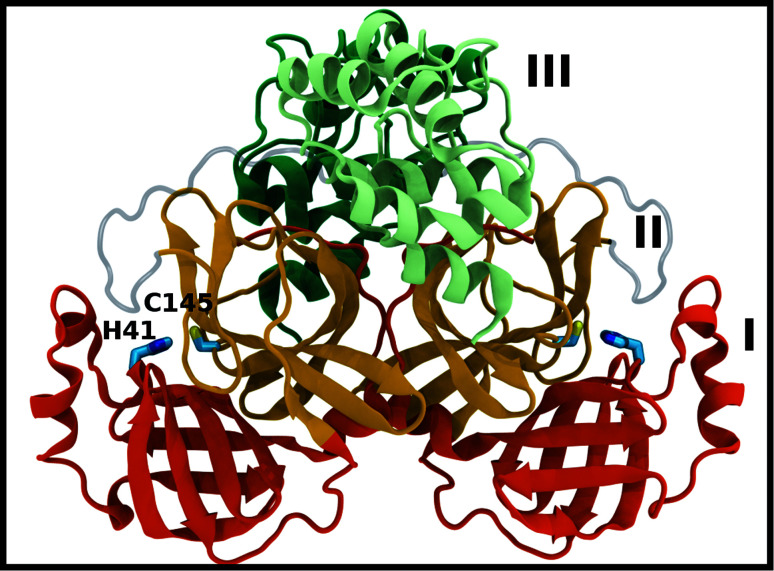
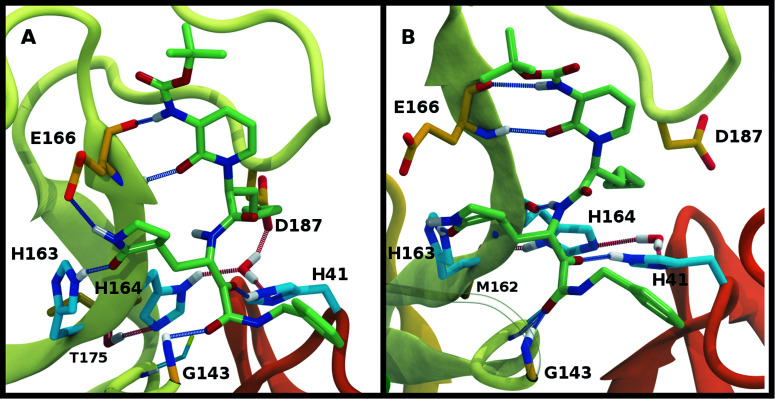
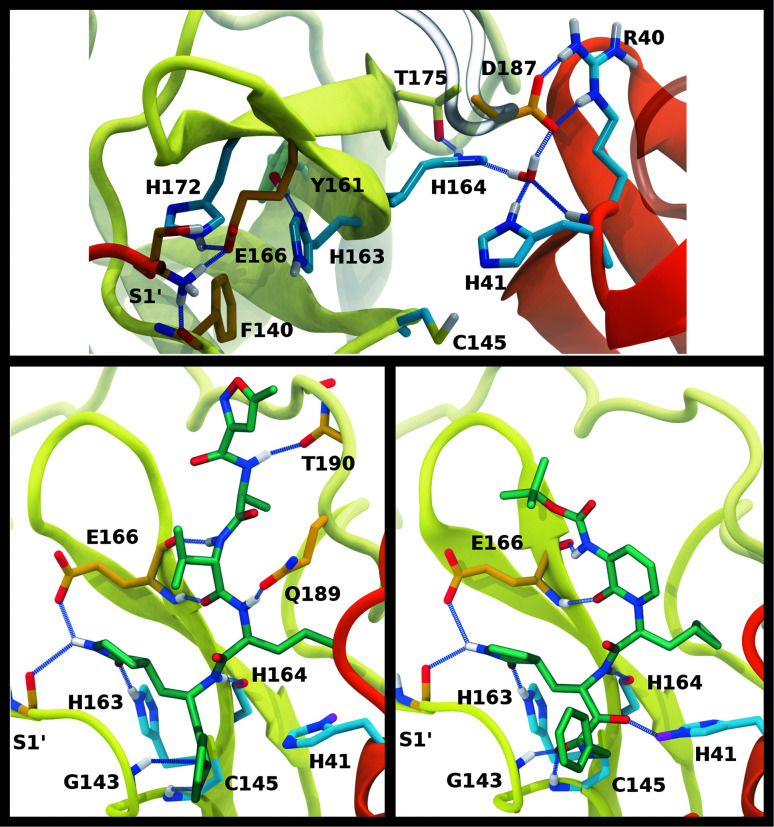
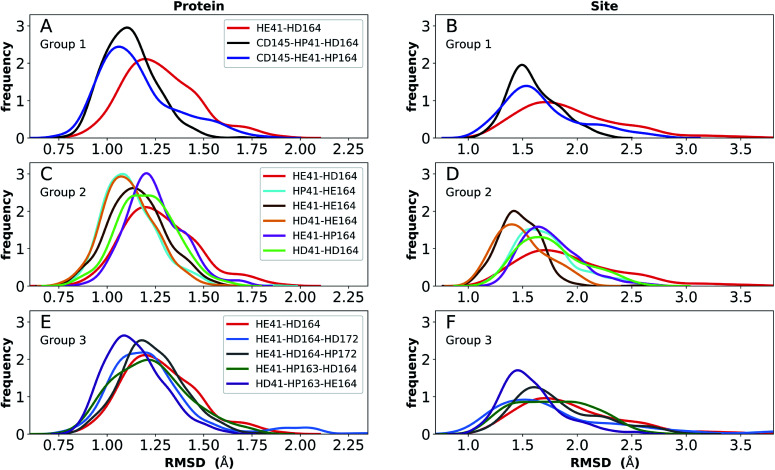
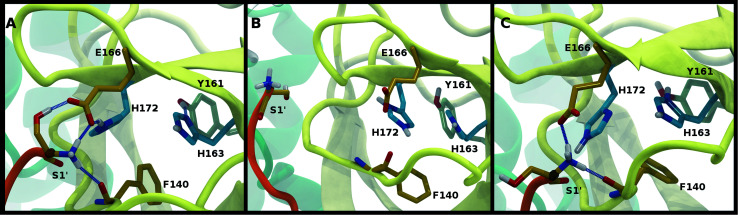
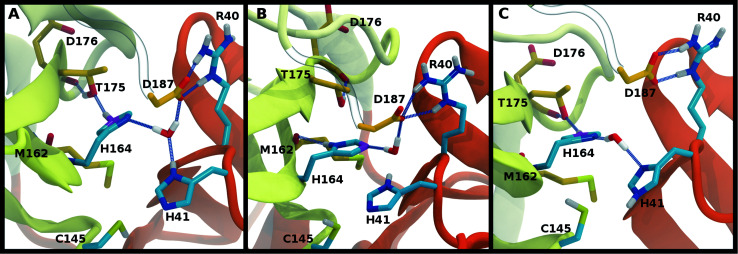
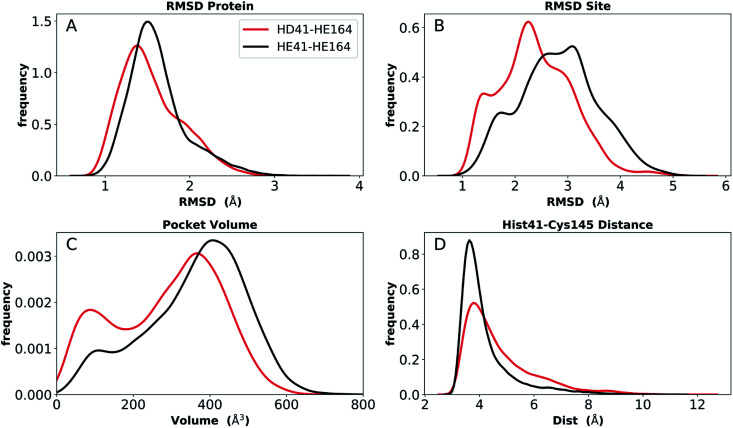
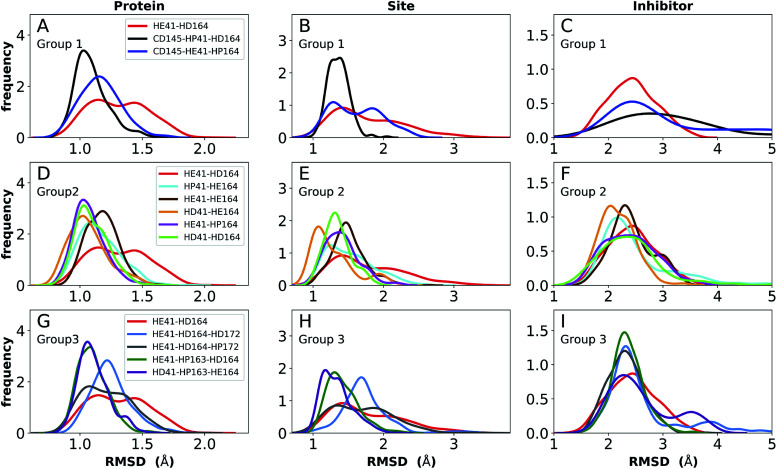
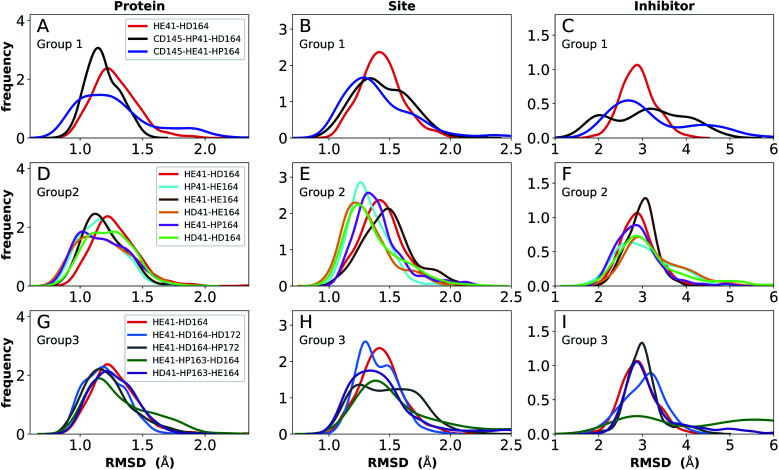
The main protease (M) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is an attractive target for antiviral therapeutics. Recently, many high-resolution apo and inhibitor-bound structures of M, a cysteine protease, have been determined, facilitating structure-based drug design. M plays a central role in the viral life cycle by catalyzing the cleavage of SARS-CoV-2 polyproteins. In addition to the catalytic dyad His41-Cys145, M contains multiple histidines including His163, His164, and His172. The protonation states of these histidines and the catalytic nucleophile Cys145 have been debated in previous studies of SARS-CoV M, but have yet to be investigated for SARS-CoV-2. In this work we have used molecular dynamics simulations to determine the structural stability of SARS-CoV-2 M as a function of the protonation assignments for these residues. We simulated both the apo and inhibitor-bound enzyme and found that the conformational stability of the binding site, bound inhibitors, and the hydrogen bond networks of M are highly sensitive to these assignments. Additionally, the two inhibitors studied, the peptidomimetic N3 and an α-ketoamide, display distinct His41/His164 protonation-state-dependent stabilities. While the apo and the N3-bound systems favored N (HD) and N (HE) protonation of His41 and His164, respectively, the α-ketoamide was not stably bound in this state. Our results illustrate the importance of using appropriate histidine protonation states to accurately model the structure and dynamics of SARS-CoV-2 M in both the apo and inhibitor-bound states, a necessary prerequisite for drug-design efforts.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)的主要蛋白酶(M)是抗病毒治疗的一个有吸引力的靶点。最近,已经确定了许多高分辨率的M(一种半胱氨酸蛋白酶)的无配体和与抑制剂结合的结构,这有助于基于结构的药物设计。M通过催化SARS-CoV-2多蛋白的切割在病毒生命周期中发挥核心作用。除了催化二元组His41-Cys145外,M还包含多个组氨酸,包括His163、His164和His172。在先前对SARS-CoV M的研究中,这些组氨酸的质子化状态以及催化亲核试剂Cys145一直存在争议,但尚未对SARS-CoV-2进行研究。在这项工作中,我们使用分子动力学模拟来确定SARS-CoV-2 M的结构稳定性与这些残基质子化分配的函数关系。我们模拟了无配体和与抑制剂结合的酶,发现结合位点、结合的抑制剂以及M的氢键网络的构象稳定性对这些分配高度敏感。此外,所研究的两种抑制剂,肽模拟物N3和一种α-酮酰胺,表现出不同的His41/His164质子化状态依赖性稳定性。虽然无配体和与N3结合的系统分别有利于His41和His164的N (HD)和N (HE)质子化,但α-酮酰胺在这种状态下结合不稳定。我们的结果说明了使用适当的组氨酸质子化状态来准确模拟SARS-CoV-2 M在无配体和与抑制剂结合状态下的结构和动力学的重要性,这是药物设计工作的必要前提。