Department of Neurology, Medical Faculty, RWTH Aachen University, 52074 Aachen, Germany.
JARA-BRAIN Institute Molecular Neuroscience and Neuroimaging, Forschungszentrum Jülich GmbH and RWTH Aachen University, 52074 Aachen, Germany.
Int J Mol Sci. 2021 Oct 28;22(21):11586. doi: 10.3390/ijms222111586.
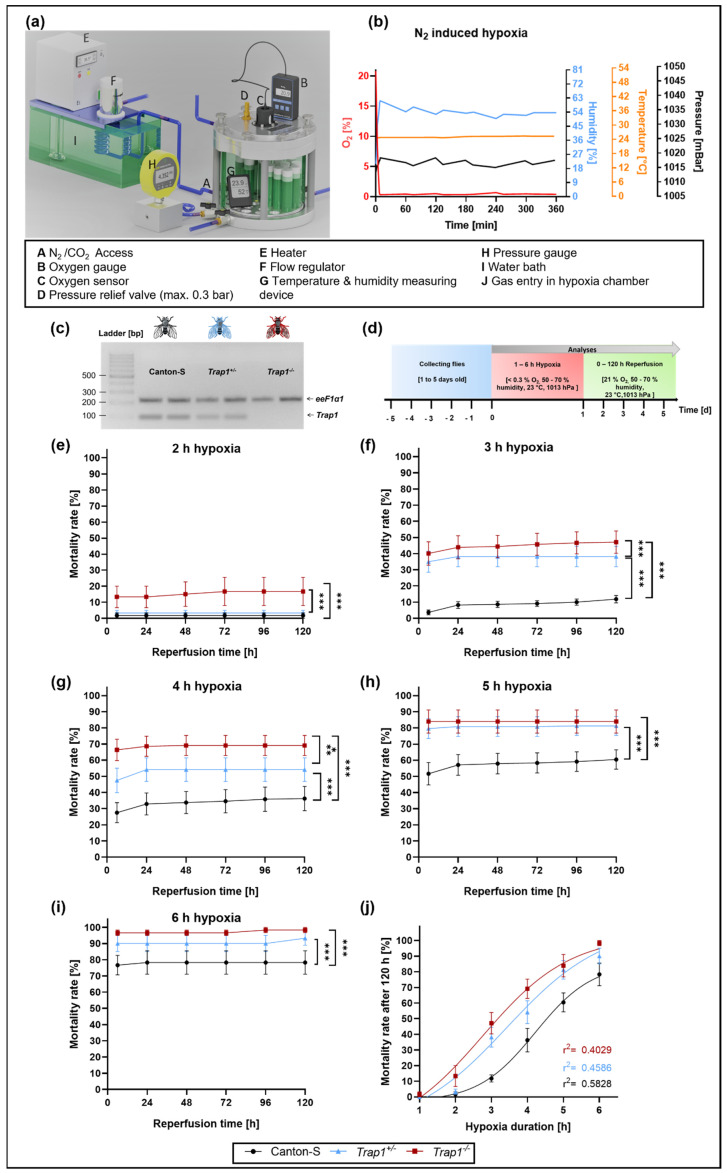
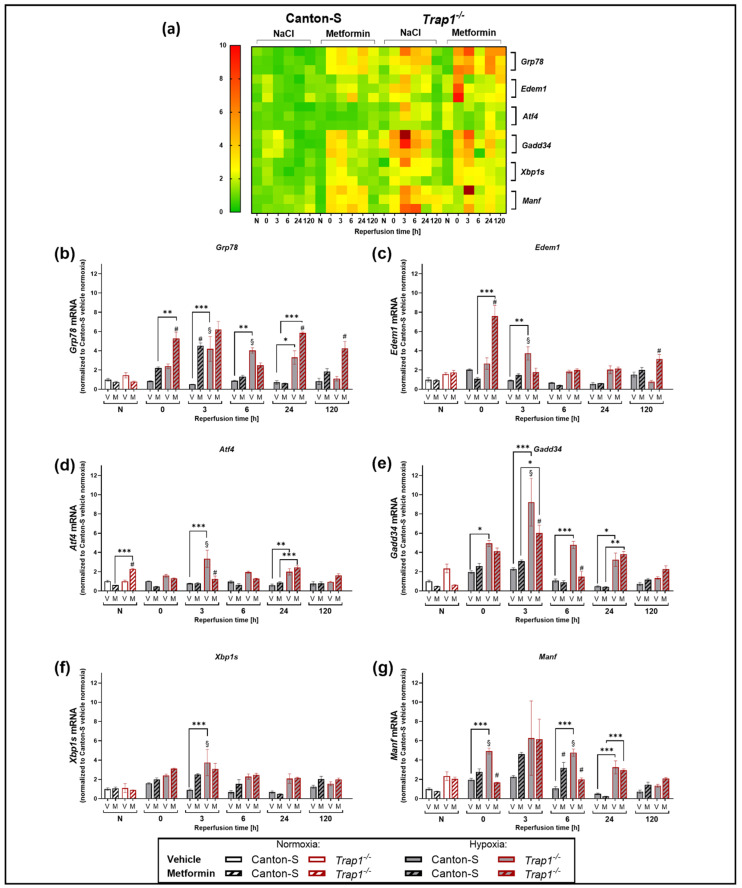
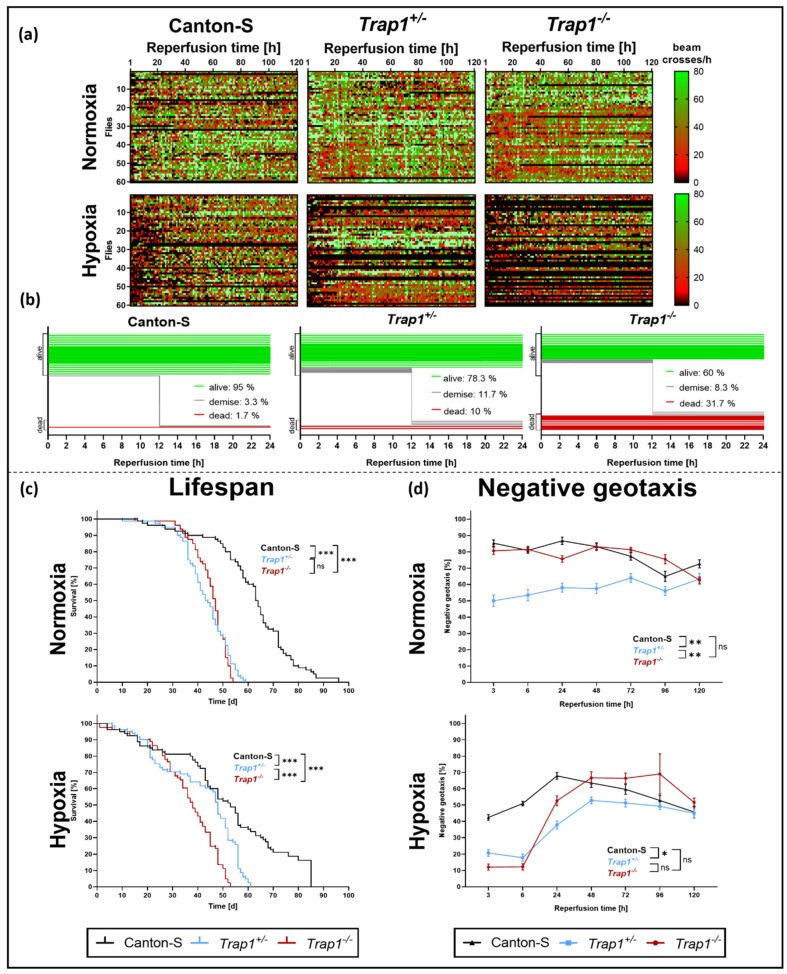
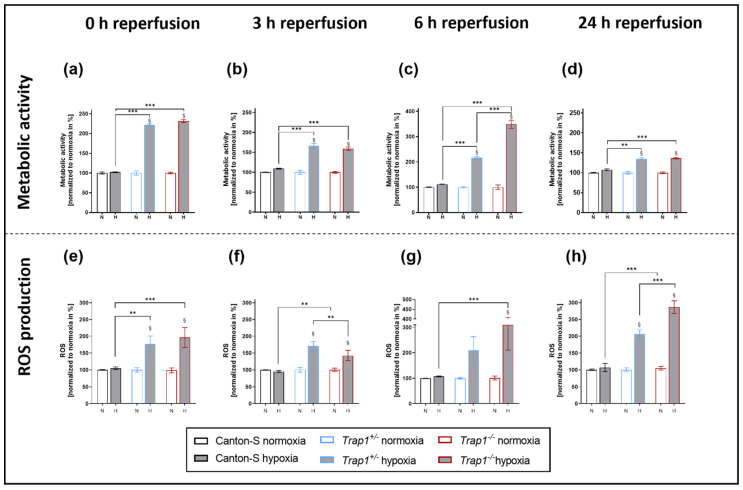
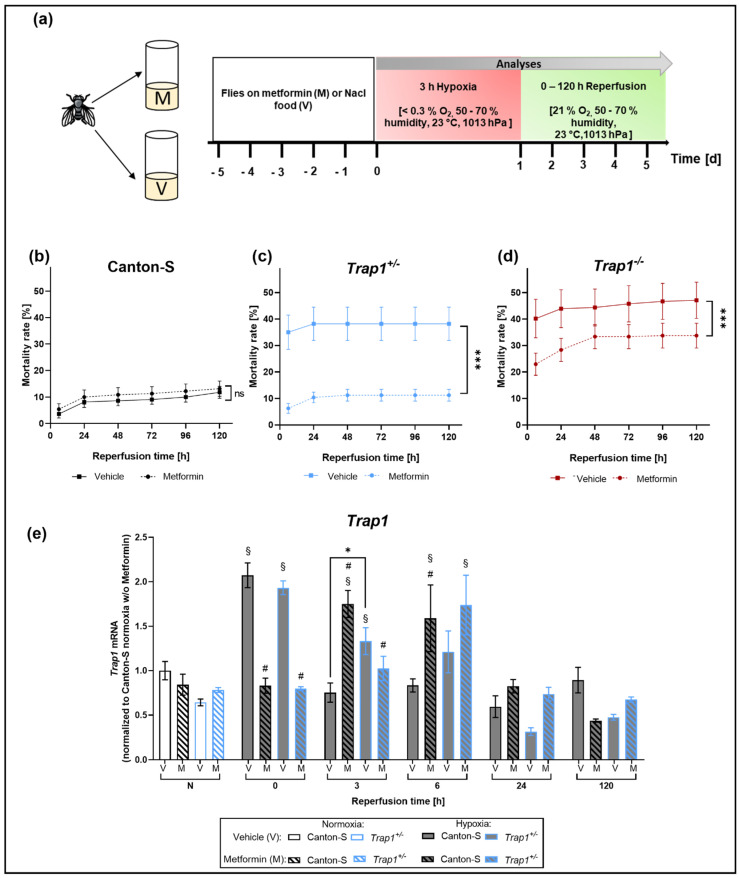
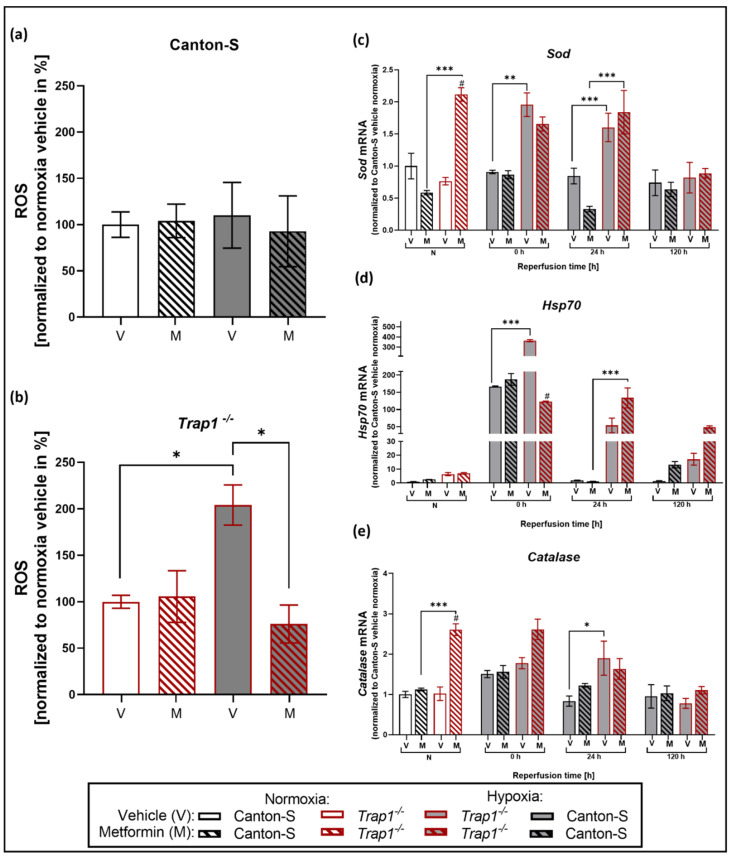
Hypoxia is known to impair mitochondrial and endoplasmic reticulum (ER) homeostasis. Post-hypoxic perturbations of the ER proteostasis result in the accumulation of misfolded/unfolded proteins leading to the activation of the Unfolded Protein Response (UPR). Mitochondrial chaperone TNF receptor-associated protein 1 (TRAP1) is reported to preserve mitochondrial membrane potential and to impede reactive oxygen species (ROS) production thereby protecting cells from ER stress as well as oxidative stress. The first-line antidiabetic drug Metformin has been attributed a neuroprotective role after hypoxia. Interestingly, Metformin has been reported to rescue mitochondrial deficits in fibroblasts derived from a patient carrying a homozygous TRAP1 loss-of-function mutation. We sought to investigate a putative link between Metformin, TRAP1, and the UPR after hypoxia. We assessed post-hypoxic/reperfusion longevity, mortality, negative geotaxis, ROS production, metabolic activity, gene expression of antioxidant proteins, and activation of the UPR in -deficient flies. Following hypoxia, deficiency caused higher mortality and greater impairments in negative geotaxis compared to controls. Similarly, post-hypoxic production of ROS and UPR activation was significantly higher in -deficient compared to control flies. Metformin counteracted the deleterious effects of hypoxia in -deficient flies but had no protective effect in wild-type flies. We provide evidence that TRAP1 is crucially involved in the post-hypoxic regulation of mitochondrial/ER stress and the activation of the UPR. Metformin appears to rescue -deficiency after hypoxia mitigating ROS production and downregulating the pro-apoptotic PERK (protein kinase R-like ER kinase) arm of the UPR.
缺氧已知会损害线粒体和内质网 (ER) 的稳态。缺氧后 ER 蛋白稳态的后扰导致错误折叠/未折叠蛋白质的积累,导致未折叠蛋白反应 (UPR) 的激活。已经报道线粒体伴侣 TNF 受体相关蛋白 1 (TRAP1) 可维持线粒体膜电位并阻止活性氧 (ROS) 的产生,从而保护细胞免受 ER 应激和氧化应激的影响。一线抗糖尿病药物二甲双胍在缺氧后被归因于神经保护作用。有趣的是,据报道二甲双胍可挽救携带 TRAP1 功能丧失突变纯合子的患者来源成纤维细胞中的线粒体缺陷。我们试图研究缺氧后二甲双胍、TRAP1 和 UPR 之间的潜在联系。我们评估了缺氧后再灌注的寿命、死亡率、负趋地性、ROS 产生、代谢活性、抗氧化蛋白的基因表达和 UPR 的激活。缺氧后,与对照相比, 缺陷导致更高的死亡率和更严重的负趋地性障碍。同样,与对照相比,缺氧后 ROS 的产生和 UPR 的激活在 -缺陷果蝇中显著增加。二甲双胍可拮抗缺氧对 -缺陷果蝇的有害影响,但对野生型果蝇没有保护作用。我们提供的证据表明 TRAP1 在内质网/线粒体应激和 UPR 激活的缺氧后调节中起关键作用。二甲双胍似乎可挽救缺氧后的 -缺陷,减轻 ROS 的产生并下调 UPR 的促凋亡 PERK (蛋白激酶 R 样内质网激酶) 臂。