Marques Andreia T, Tanoeiro Luís, Duarte Aida, Gonçalves Luisa, Vítor Jorge M B, Vale Filipa F
Pathogen Genome Bioinformatics and Computational Biology, Research Institute for Medicines (iMed-ULisboa), Faculty of Pharmacy, Universidade de Lisboa, 1649-003 Lisboa, Portugal.
Faculty of Pharmacy, Universidade de Lisboa, Av. Gama Pinto, 1649-003 Lisboa, Portugal.
Microorganisms. 2021 Oct 28;9(11):2252. doi: 10.3390/microorganisms9112252.
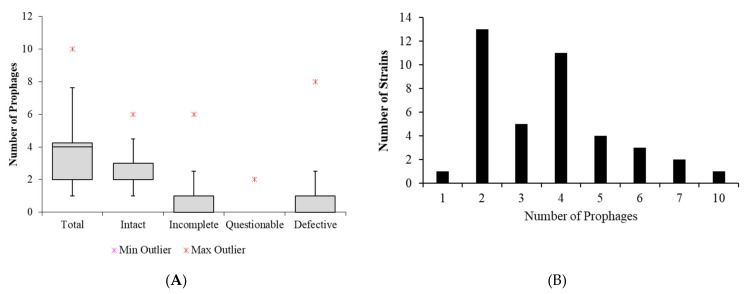
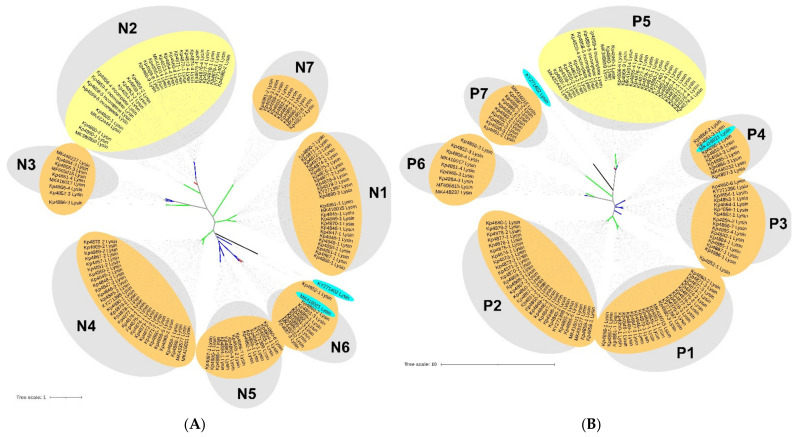
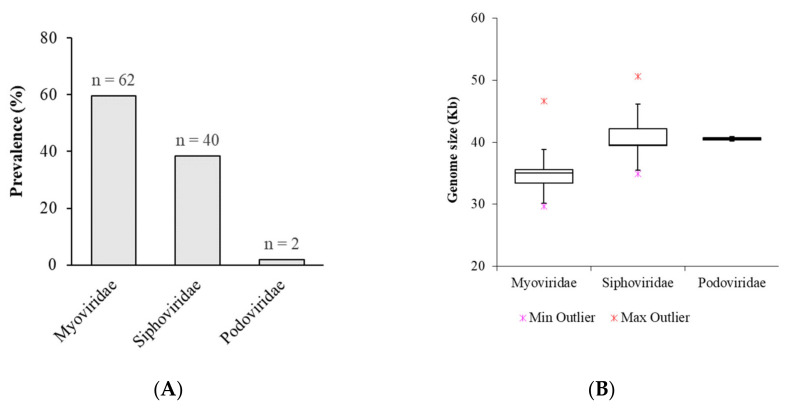
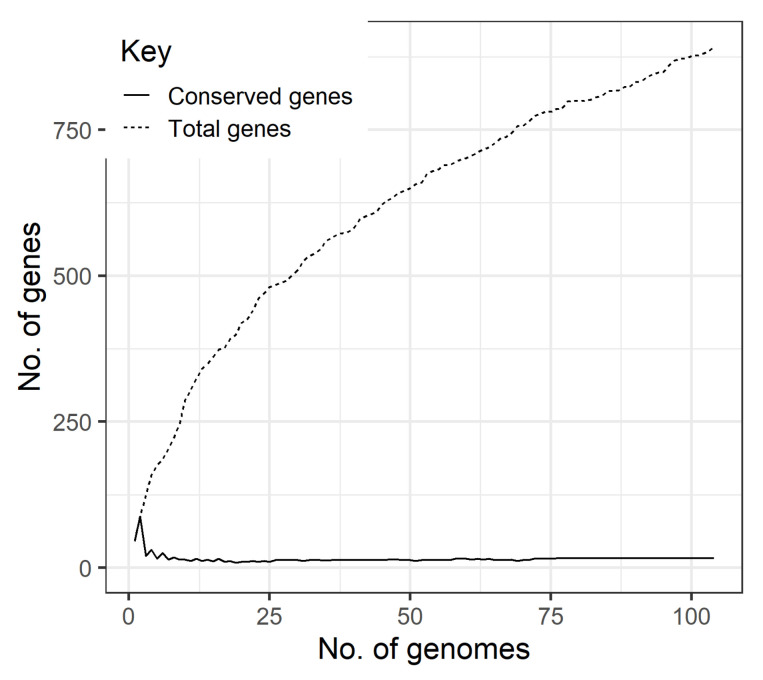
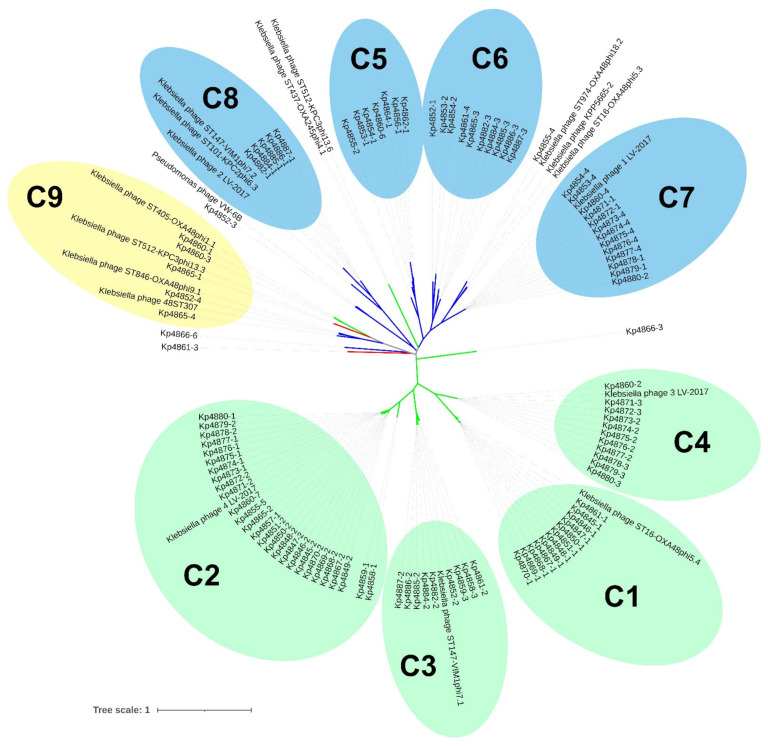
is an increasing threat to public health and represents one of the most concerning pathogens involved in life-threatening infections. The resistant and virulence determinants are coded by mobile genetic elements which can easily spread between bacteria populations and co-evolve with its genomic host. In this study, we present the full genomic sequences, insertion sites and phylogenetic analysis of 150 prophages found in 40 clinical isolates obtained from an outbreak in a Portuguese hospital. All strains harbored at least one prophage and we identified 104 intact prophages (69.3%). The prophage size ranges from 29.7 to 50.6 kbp, coding between 32 and 78 putative genes. The prophage GC content is 51.2%, lower than the average GC content of 57.1% in . Complete prophages were classified into three families in the order : (59.6%), (38.5%) and (1.9%). In addition, an alignment and phylogenetic analysis revealed nine distinct clusters. Evidence of recombination was detected within the genome of some prophages but, in most cases, proteins involved in viral structure, transcription, replication and regulation (lysogenic/lysis) were maintained. These results support the knowledge that prophages are diverse and widely disseminated in genomes, contributing to the evolution of this species and conferring additional phenotypes. Moreover, we identified prophages in a set of endolysin genes, which were found to code for proteins with lysozyme activity, cleaving the β-1,4 linkages between N-acetylmuramic acid and N-acetyl-D-glucosamine residues in the peptidoglycan network and thus representing genes with the potential for lysin phage therapy.
对公共卫生构成日益严重的威胁,是涉及危及生命感染的最令人担忧的病原体之一。耐药性和毒力决定因素由可移动遗传元件编码,这些元件可在细菌群体之间轻松传播并与其基因组宿主共同进化。在本研究中,我们展示了从葡萄牙一家医院爆发疫情中获得的40株临床分离株中发现的150个原噬菌体的完整基因组序列、插入位点和系统发育分析。所有菌株都至少含有一个原噬菌体,我们鉴定出104个完整的原噬菌体(69.3%)。原噬菌体大小范围从29.7到50.6千碱基对,编码32到78个推定基因。原噬菌体的GC含量为51.2%,低于[此处原文缺失具体比较对象]平均57.%.的GC含量。完整的原噬菌体按顺序分为三个家族:此处原文缺失具体家族名称、此处原文缺失具体家族名称和此处原文缺失具体家族名称。此外,比对和系统发育分析揭示了九个不同的簇。在一些原噬菌体的基因组中检测到重组证据,但在大多数情况下,参与病毒结构、转录、复制和调控(溶原性/裂解)的蛋白质得以保留。这些结果支持了这样的认识,即原噬菌体在[此处原文缺失具体物种名称]基因组中多样且广泛传播,有助于该物种的进化并赋予额外的表型。此外,我们在一组内溶素基因中鉴定出原噬菌体,发现它们编码具有溶菌酶活性的蛋白质,可切割肽聚糖网络中N - 乙酰胞壁酸和N - 乙酰 - D - 葡萄糖胺残基之间的β - 1,4连接,因此代表具有溶菌素噬菌体治疗潜力的基因。