Division of Biomedical Convergence, College of Biomedical Science, Institute of Bioscience & Biotechnology, Kangwon National University, Chuncheon, 24341, Republic of Korea.
GENINUS Inc., Seoul, 05836, Republic of Korea.
J Transl Med. 2021 Nov 29;19(1):485. doi: 10.1186/s12967-021-03154-0.
Comparing the microbiome compositions obtained under different physiological conditions has frequently been attempted in recent years to understand the functional influence of microbiomes in the occurrence of various human diseases.
In the present work, we analyzed 102 microbiome datasets containing tumor- and normal tissue-derived microbiomes obtained from a total of 51 Korean colorectal cancer (CRC) patients using 16S rRNA amplicon sequencing. Two types of comparisons were used: 'normal versus (vs.) tumor' comparison and 'recurrent vs. nonrecurrent' comparison, for which the prognosis of patients was retrospectively determined.
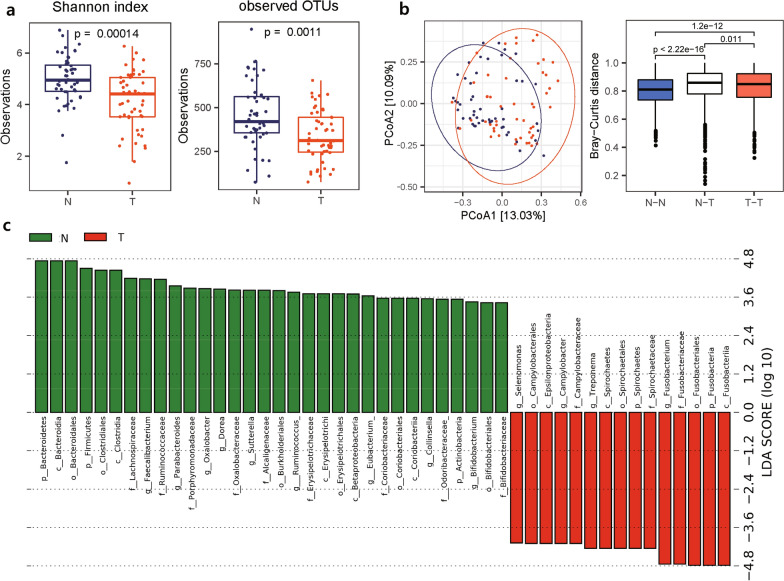
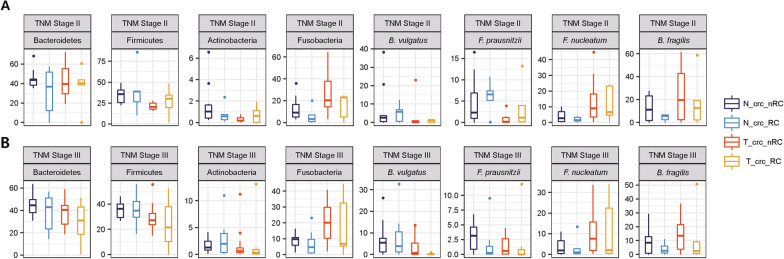
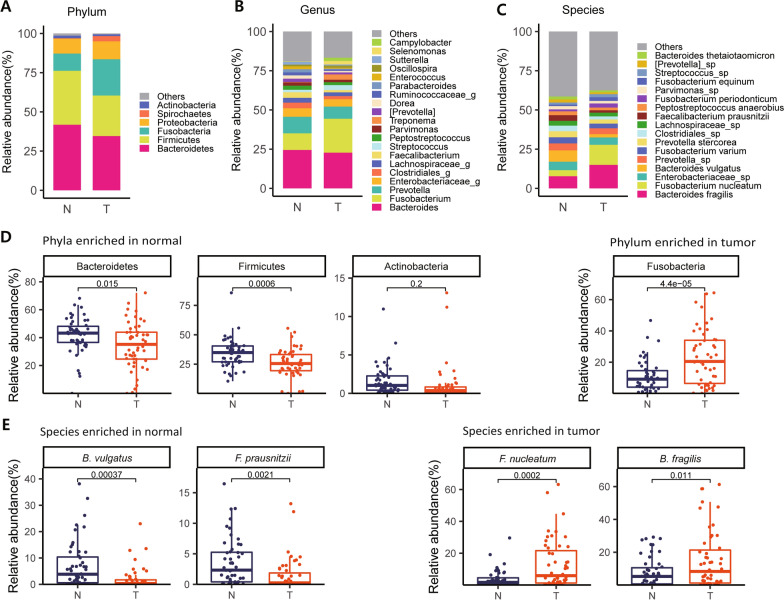
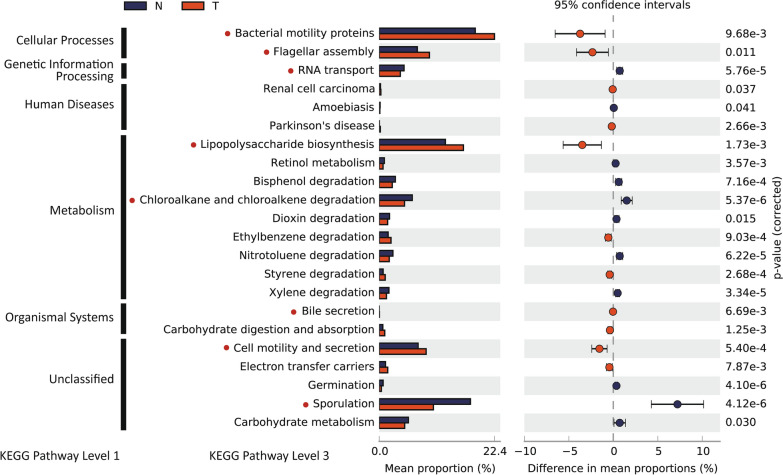
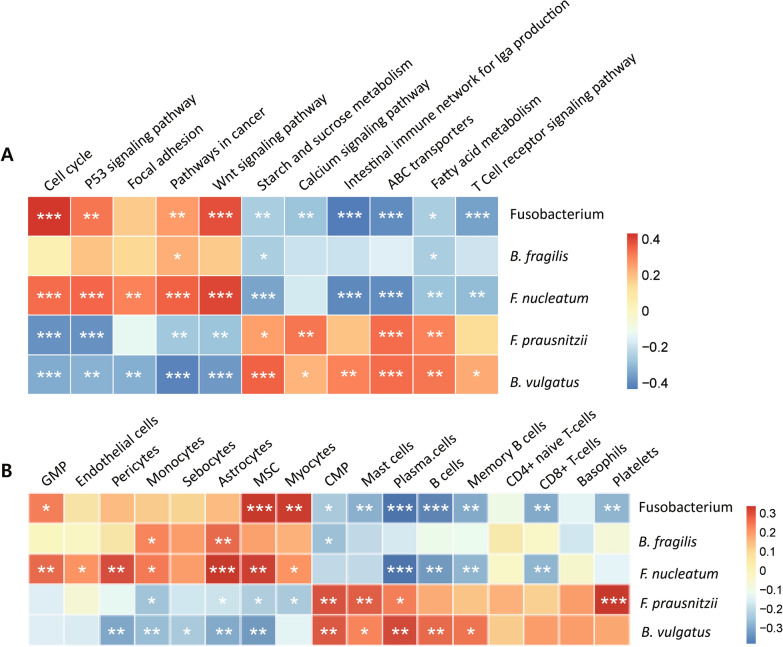
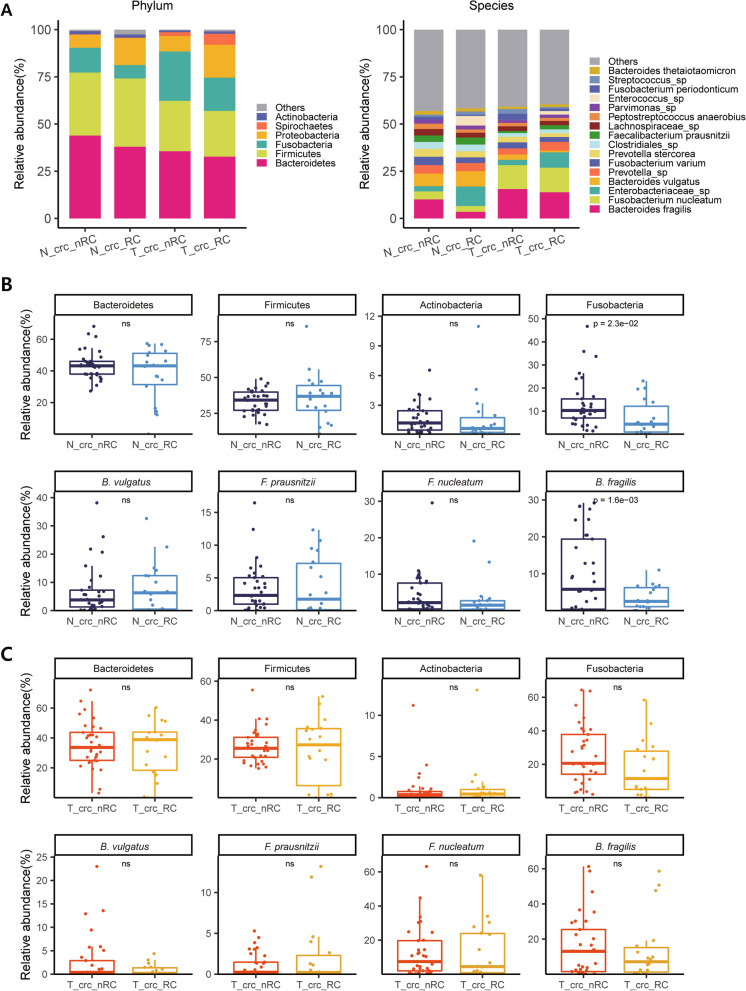
As a result, we observed that in the 'normal vs. tumor' comparison, three phyla, Firmicutes, Actinobacteria, and Bacteroidetes, were more abundant in normal tissues, whereas some pathogenic bacteria, including Fusobacterium nucleatum and Bacteroides fragilis, were more abundant in tumor tissues. We also found that bacteria with metabolic pathways related to the production of bacterial motility proteins or bile acid secretion were more enriched in tumor tissues. In addition, the amount of these two pathogenic bacteria was positively correlated with the expression levels of host genes involved in the cell cycle and cell proliferation, confirming the association of microbiomes with tumorigenic pathway genes in the host. Surprisingly, in the 'recurrent vs. nonrecurrent' comparison, we observed that these two pathogenic bacteria were more abundant in the patients without recurrence than in the patients with recurrence. The same conclusion was drawn in the analysis of both normal and tumor-derived microbiomes.
Taken together, it seems that understanding the composition of tissue microbiomes is useful for predicting the prognosis of CRC patients.
近年来,人们经常尝试比较不同生理条件下获得的微生物组组成,以了解微生物组在各种人类疾病发生中的功能影响。
在本工作中,我们使用 16S rRNA 扩增子测序分析了 102 个微生物组数据集,这些数据集包含来自 51 名韩国结直肠癌 (CRC) 患者的肿瘤和正常组织来源的微生物组。我们进行了两种类型的比较:“正常与肿瘤”比较和“复发与非复发”比较,其中患者的预后是回顾性确定的。
结果表明,在“正常与肿瘤”比较中,厚壁菌门、放线菌门和拟杆菌门在正常组织中更为丰富,而一些病原菌,包括具核梭杆菌和脆弱拟杆菌,在肿瘤组织中更为丰富。我们还发现与细菌运动蛋白产生或胆汁酸分泌相关的代谢途径的细菌在肿瘤组织中更为丰富。此外,这两种病原菌的数量与参与细胞周期和细胞增殖的宿主基因的表达水平呈正相关,证实了微生物组与宿主肿瘤发生途径基因的关联。令人惊讶的是,在“复发与非复发”比较中,我们观察到这两种病原菌在无复发患者中的丰度高于复发患者。在正常和肿瘤衍生微生物组的分析中得出了相同的结论。
总之,了解组织微生物组的组成似乎有助于预测 CRC 患者的预后。