Tsutakawa Susan E, Bacolla Albino, Katsonis Panagiotis, Bralić Amer, Hamdan Samir M, Lichtarge Olivier, Tainer John A, Tsai Chi-Lin
Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA, United States.
Department of Molecular and Cellular Oncology, University of Texas M.D. Anderson Cancer Center, Houston, TX, United States.
Front Mol Biosci. 2021 Dec 14;8:791792. doi: 10.3389/fmolb.2021.791792. eCollection 2021.
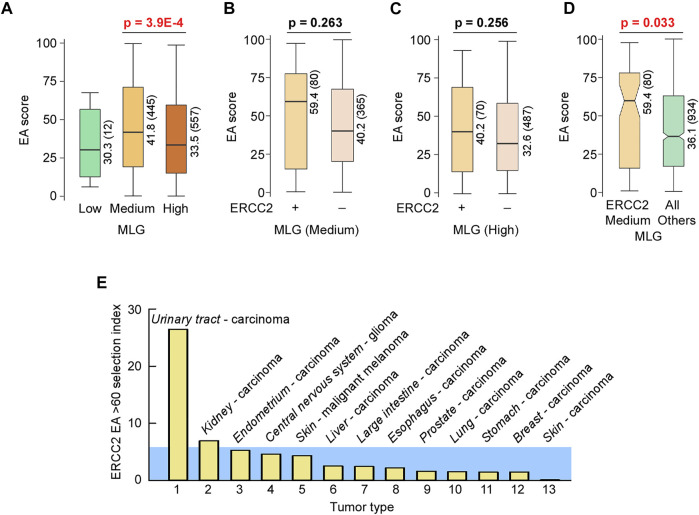
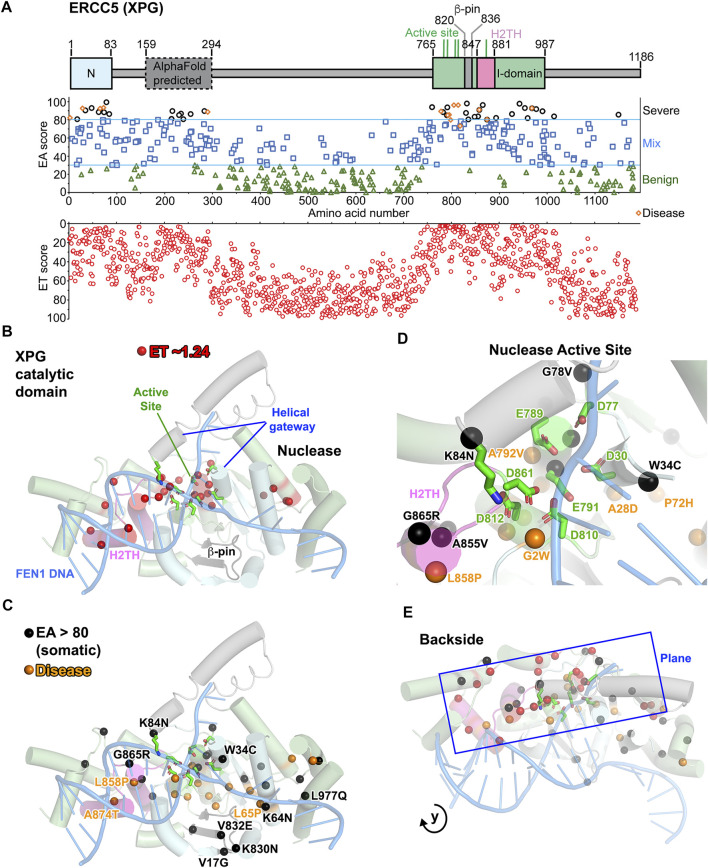
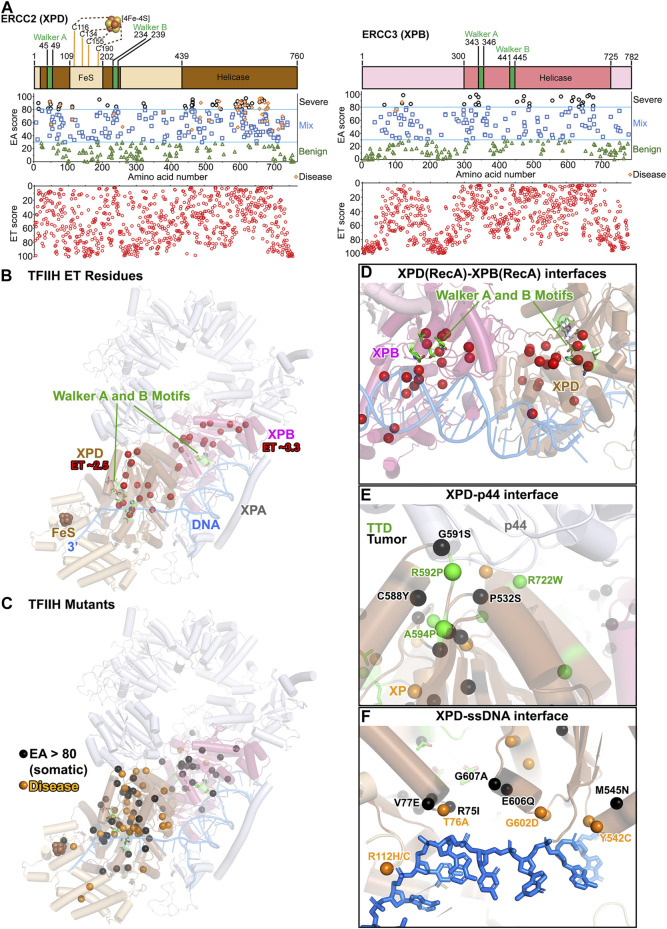
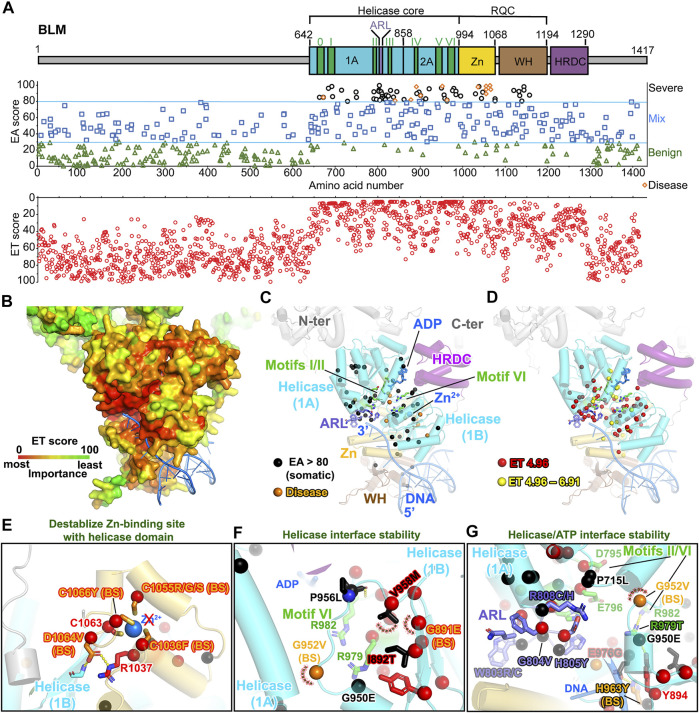
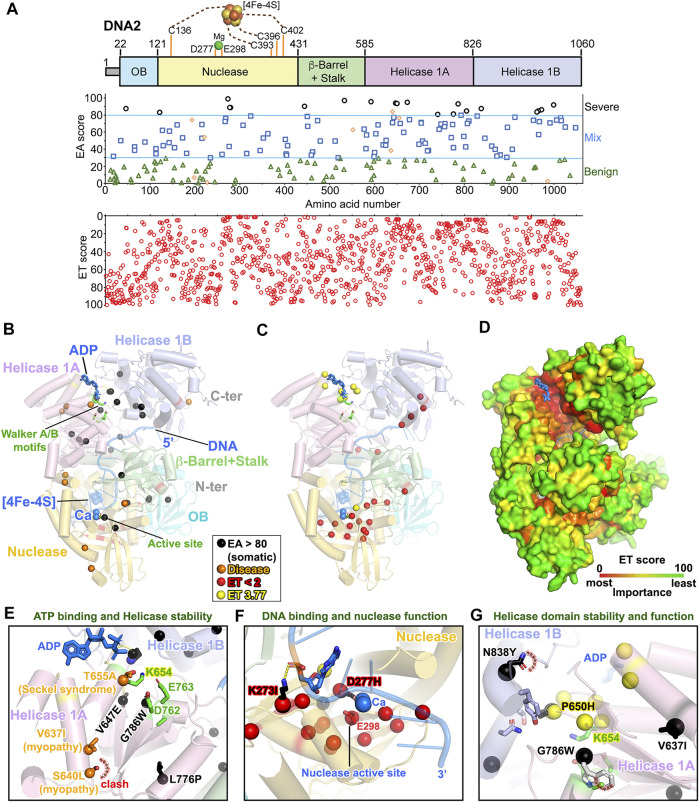
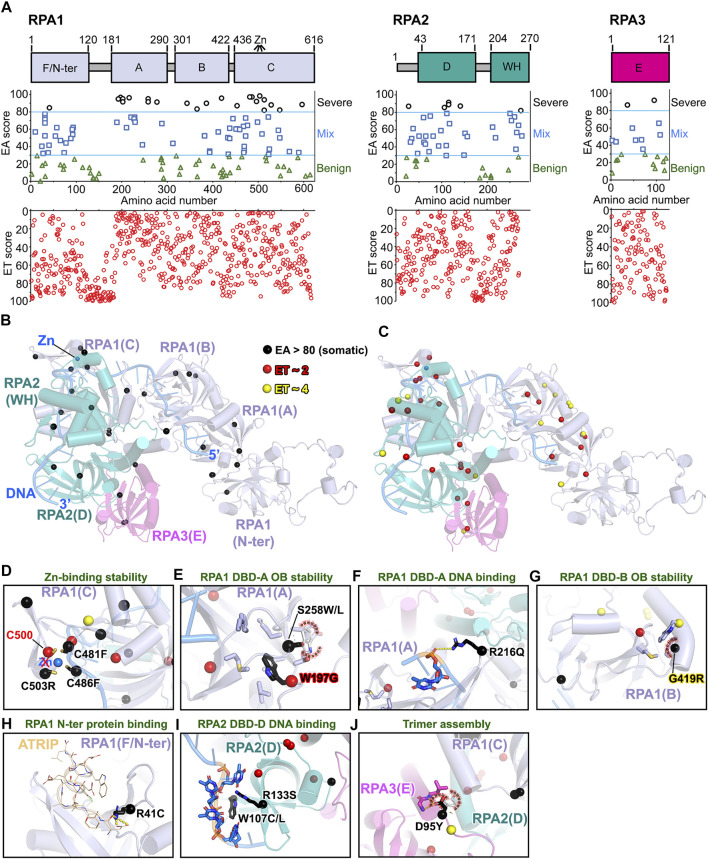
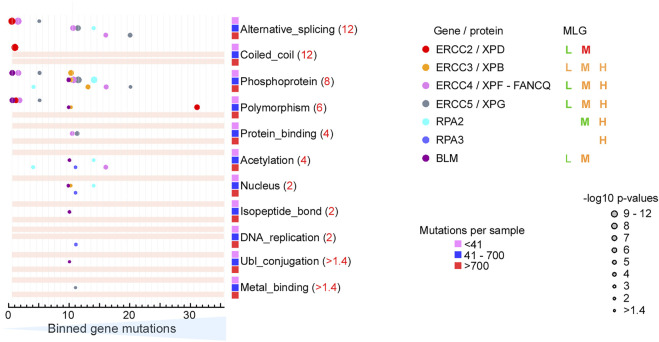
All tumors have DNA mutations, and a predictive understanding of those mutations could inform clinical treatments. However, 40% of the mutations are variants of unknown significance (VUS), with the challenge being to objectively predict whether a VUS is pathogenic and supports the tumor or whether it is benign. To objectively decode VUS, we mapped cancer sequence data and evolutionary trace (ET) scores onto crystallography and cryo-electron microscopy structures with variant impacts quantitated by evolutionary action (EA) measures. As tumors depend on helicases and nucleases to deal with transcription/replication stress, we targeted helicase-nuclease-RPA complexes: (1) XPB-XPD (within TFIIH), XPF-ERCC1, XPG, and RPA for transcription and nucleotide excision repair pathways and (2) BLM, EXO5, and RPA plus DNA2 for stalled replication fork restart. As validation, EA scoring predicts severe effects for most disease mutations, but disease mutants with low ET scores not only are likely destabilizing but also disrupt sophisticated allosteric mechanisms. For sites of disease mutations and VUS predicted to be severe, we found strong co-localization to ordered regions. Rare discrepancies highlighted the different survival requirements between disease and tumor mutations, as well as the value of examining proteins within complexes. In a genome-wide analysis of 33 cancer types, we found correlation between the number of mutations in each tumor and which pathways or functional processes in which the mutations occur, revealing different mutagenic routes to tumorigenesis. We also found upregulation of ancient genes including BLM, which supports a non-random and concerted cancer process: reversion to a unicellular, proliferation-uncontrolled, status by breaking multicellular constraints on cell division. Together, these genes and global analyses challenge the binary "driver" and "passenger" mutation paradigm, support a gradient impact as revealed by EA scoring from moderate to severe at a single gene level, and indicate reduced regulation as well as activity. The objective quantitative assessment of VUS scoring and gene overexpression in the context of functional interactions and pathways provides insights for biology, oncology, and precision medicine.
所有肿瘤都存在DNA突变,对这些突变进行预测性了解可为临床治疗提供依据。然而,40%的突变是意义未明的变异(VUS),挑战在于客观预测一个VUS是否具有致病性并支持肿瘤生长,或者它是否是良性的。为了客观地解读VUS,我们将癌症序列数据和进化踪迹(ET)分数映射到晶体学和冷冻电子显微镜结构上,通过进化作用(EA)测量来量化变异的影响。由于肿瘤依赖解旋酶和核酸酶来应对转录/复制压力,我们将目标锁定在解旋酶-核酸酶-RPA复合物上:(1)用于转录和核苷酸切除修复途径的XPB-XPD(在TFIIH内)、XPF-ERCC1、XPG和RPA,以及(2)用于停滞复制叉重启的BLM、EXO5和RPA加DNA2。作为验证,EA评分预测大多数疾病突变会产生严重影响,但ET分数低的疾病突变体不仅可能会破坏稳定性,还会扰乱复杂的变构机制。对于预测为严重的疾病突变位点和VUS,我们发现它们与有序区域有很强的共定位。罕见的差异突出了疾病突变和肿瘤突变之间不同的生存需求,以及在复合物中检测蛋白质的价值。在对33种癌症类型进行的全基因组分析中,我们发现每个肿瘤中的突变数量与突变发生的途径或功能过程之间存在相关性,揭示了肿瘤发生的不同诱变途径。我们还发现包括BLM在内的古老基因上调,这支持了一个非随机且协同的癌症过程:通过打破对细胞分裂的多细胞限制,恢复到单细胞、不受控制的增殖状态。总之,这些基因和全局分析挑战了二元的“驱动”和“乘客”突变范式,支持了EA评分在单个基因水平上从中度到重度所揭示的梯度影响,并表明调控和活性降低。在功能相互作用和途径的背景下对VUS评分和基因过表达进行客观定量评估,为生物学、肿瘤学和精准医学提供了见解。