Mathematics and Computer Science Department, Faculty of Science, Menoufia University, Al Minufya, Egypt.
Department of Computer Science, College of Computer and Information Sciences, King Saud University, Riyadh 11543, Saudi Arabia.
Comput Intell Neurosci. 2021 Dec 29;2021:7231126. doi: 10.1155/2021/7231126. eCollection 2021.
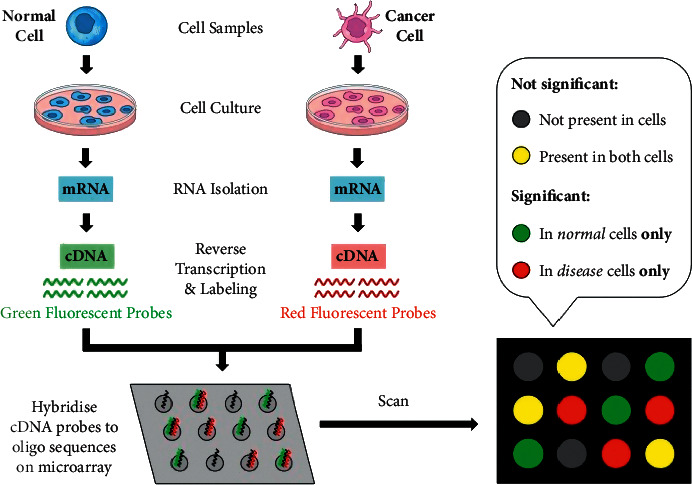
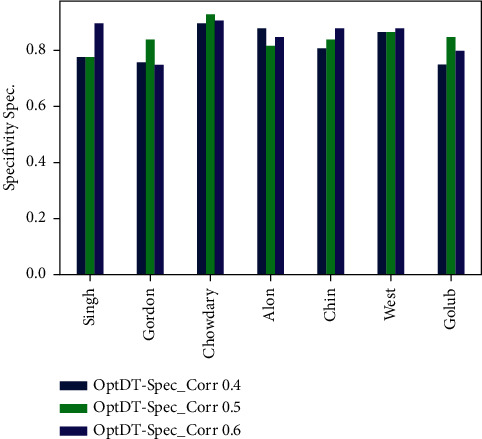
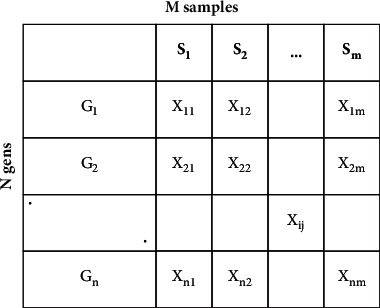
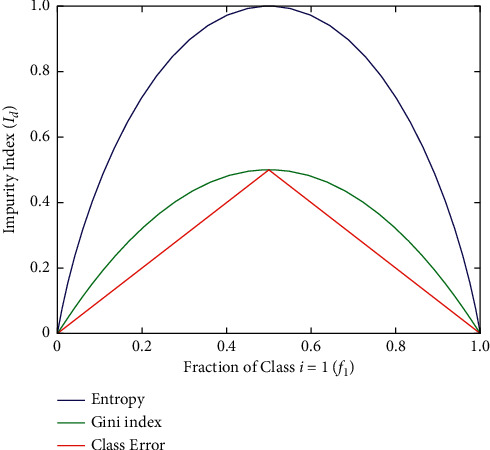
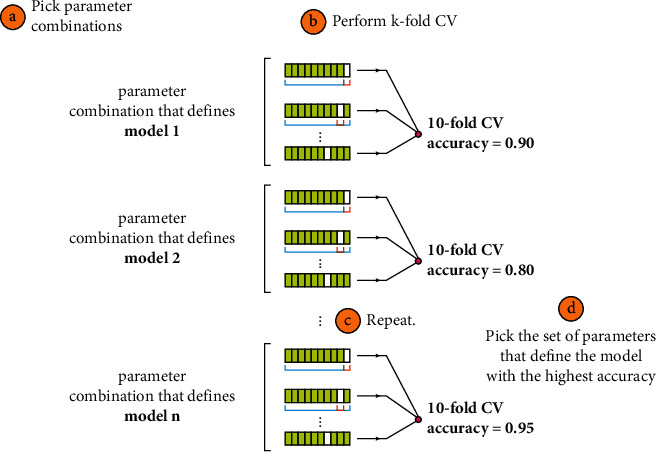
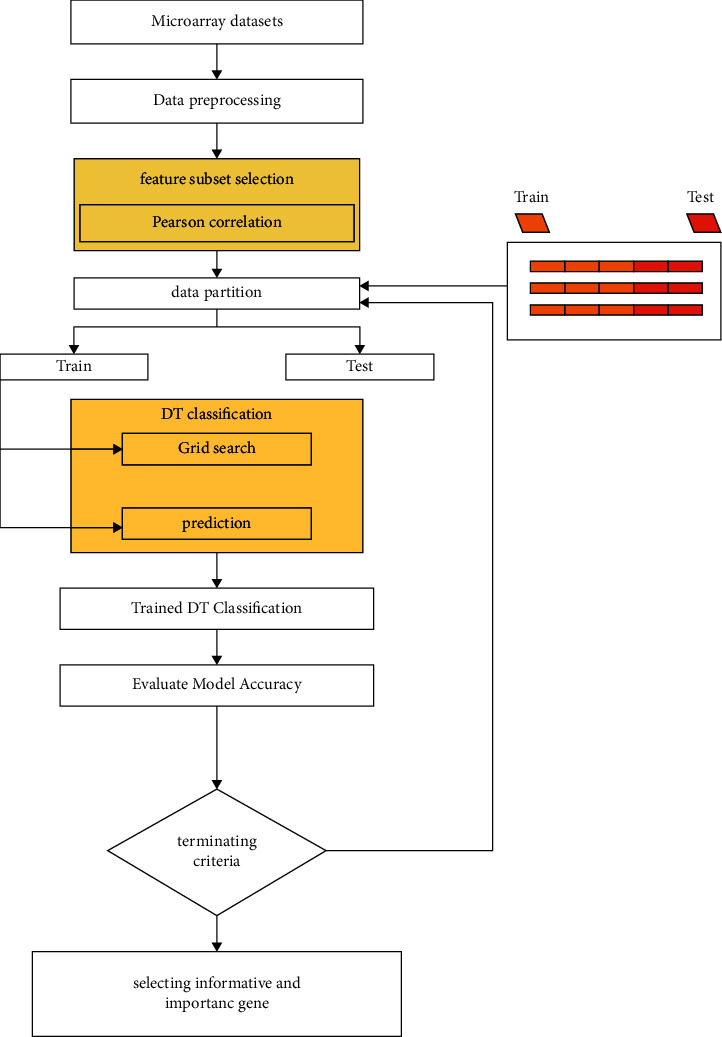
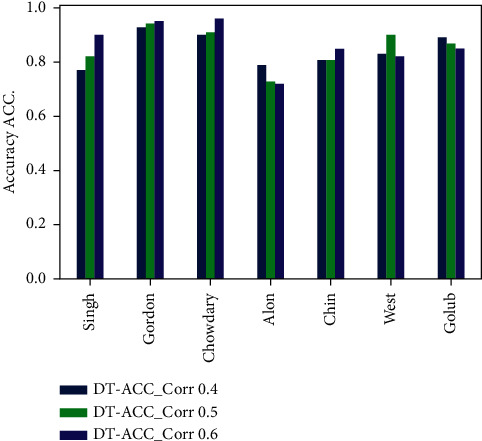
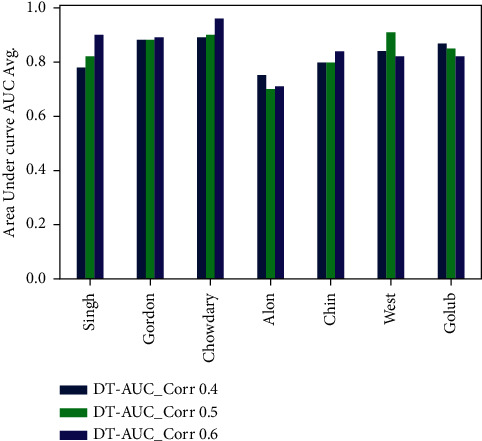
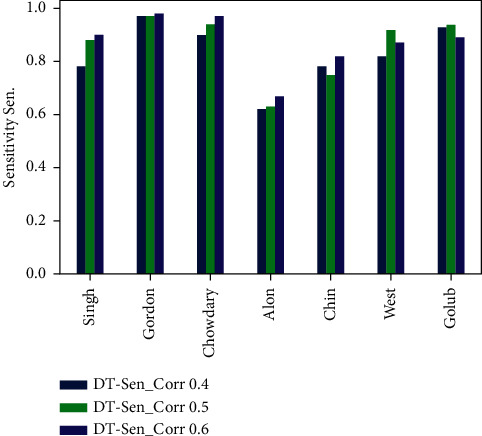
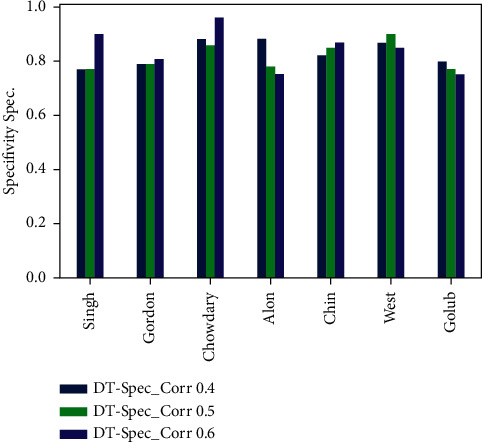
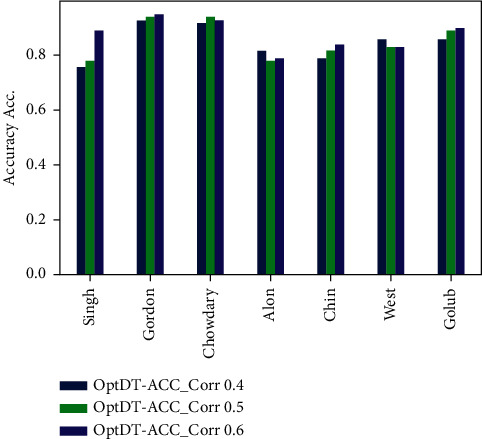
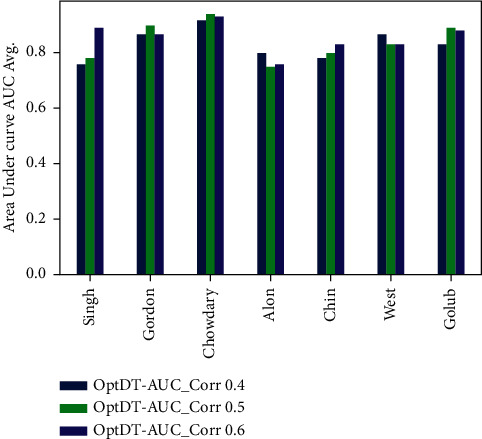
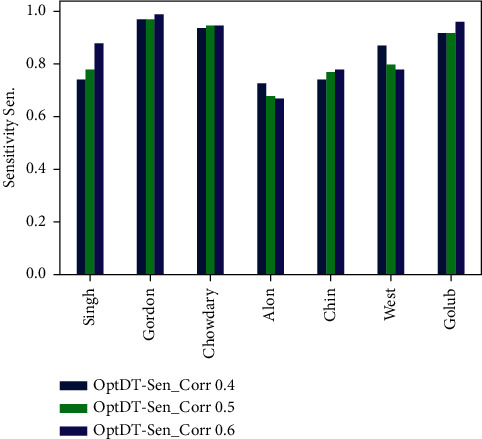
Cancer can be considered as one of the leading causes of death widely. One of the most effective tools to be able to handle cancer diagnosis, prognosis, and treatment is by using expression profiling technique which is based on microarray gene. For each data point (sample), gene data expression usually receives tens of thousands of genes. As a result, this data is large-scale, high-dimensional, and highly redundant. The classification of gene expression profiles is considered to be a (NP)-Hard problem. Feature (gene) selection is one of the most effective methods to handle this problem. A hybrid cancer classification approach is presented in this paper, and several machine learning techniques were used in the hybrid model: Pearson's correlation coefficient as a correlation-based feature selector and reducer, a Decision Tree classifier that is easy to interpret and does not require a parameter, and Grid Search CV (cross-validation) to optimize the maximum depth hyperparameter. Seven standard microarray cancer datasets are used to evaluate our model. To identify which features are the most informative and relative using the proposed model, various performance measurements are employed, including classification accuracy, specificity, sensitivity, 1-score, and AUC. The suggested strategy greatly decreases the number of genes required for classification, selects the most informative features, and increases classification accuracy, according to the results.
癌症被广泛认为是主要死因之一。为了能够进行癌症诊断、预后和治疗,使用基于微阵列基因的表达谱技术是最有效的工具之一。对于每个数据点(样本),基因数据表达通常会接收数万种基因。因此,这些数据具有大规模、高维度和高度冗余的特点。基因表达谱的分类被认为是一个(NP)-Hard 问题。特征(基因)选择是处理这个问题的最有效方法之一。本文提出了一种混合癌症分类方法,并在混合模型中使用了几种机器学习技术:Pearson 相关系数作为基于相关性的特征选择器和降维器、易于解释且不需要参数的决策树分类器,以及网格搜索 CV(交叉验证)来优化最大深度超参数。使用七种标准的微阵列癌症数据集来评估我们的模型。为了使用所提出的模型识别哪些特征是最具信息量和相关性的,使用了各种性能测量方法,包括分类准确性、特异性、敏感性、1 分数和 AUC。根据结果,该策略大大减少了分类所需的基因数量,选择了最具信息量的特征,并提高了分类准确性。