Benediktsson Bardi, Bjornsson Ragnar
Science Institute, University of Iceland, Dunhagi 3, 107 Reykjavik, Iceland.
Max-Planck Institute for Chemical Energy Conversion, Stiftstrasse 34-36, 45470 Mülheim an der Ruhr, Germany.
Inorg Chem. 2020 Aug 17;59(16):11514-11527. doi: 10.1021/acs.inorgchem.0c01320. Epub 2020 Aug 5.
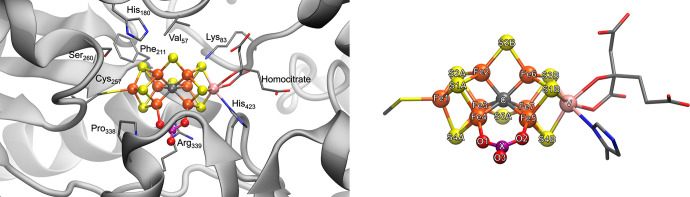
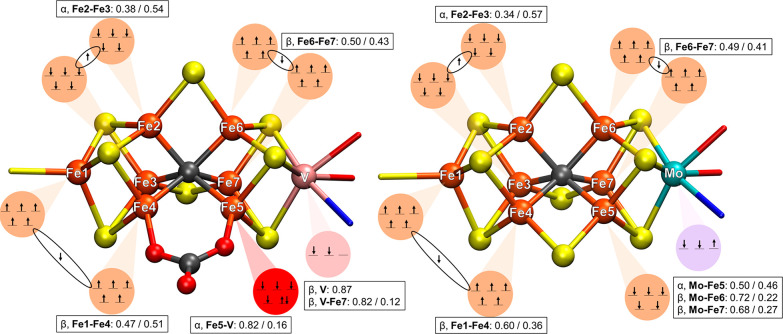
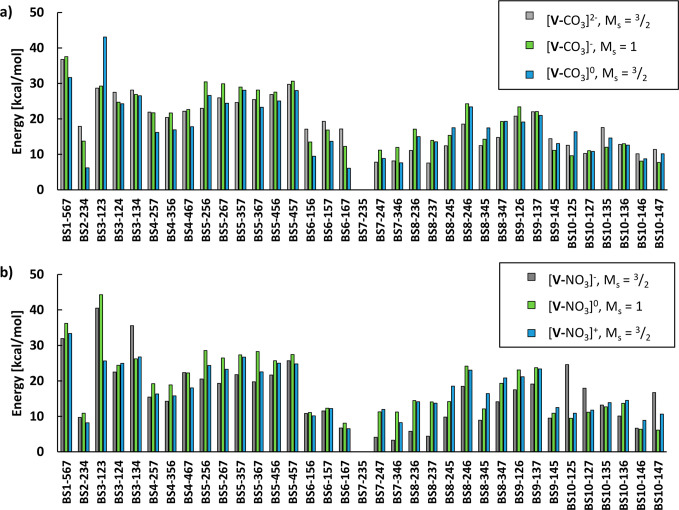
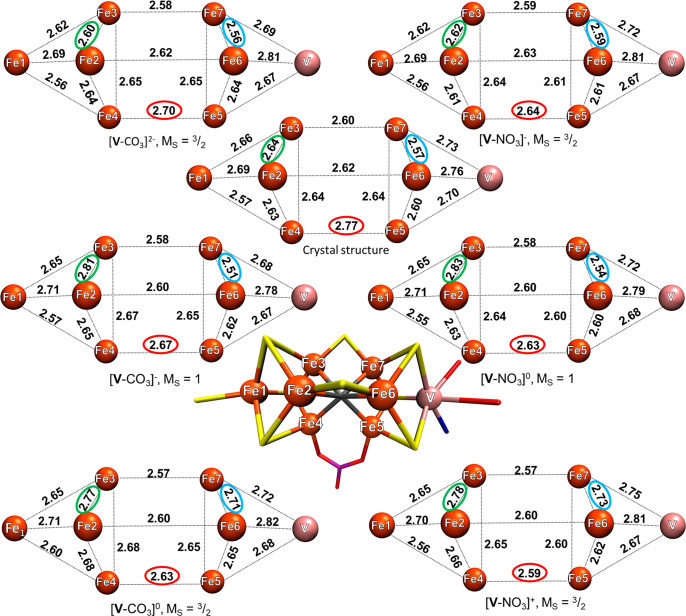
The nitrogenase enzymes are responsible for all biological nitrogen reduction. How this is accomplished at the atomic level, however, has still not been established. The molybdenum-dependent nitrogenase has been extensively studied and is the most active catalyst for dinitrogen reduction of the nitrogenase enzymes. The vanadium-dependent form, on the other hand, displays different reactivity, being capable of CO and CO reduction to hydrocarbons. Only recently did a crystal structure of the VFe protein of vanadium nitrogenase become available, paving the way for detailed theoretical studies of the iron-vanadium cofactor (FeVco) within the protein matrix. The crystal structure revealed a bridging 4-atom ligand between two Fe atoms, proposed to be either a CO or NO ligand. Using a quantum mechanics/molecular mechanics model of the VFe protein, starting from the 1.35 Å crystal structure, we have systematically explored multiple computational models for FeVco, considering either a CO or NO ligand, three different redox states, and multiple broken-symmetry states. We find that only a [VFeSC(CO)] model for FeVco reproduces the crystal structure of FeVco well, as seen in a comparison of the Fe-Fe and V-Fe distances in the computed models. Furthermore, a broken-symmetry solution with Fe2, Fe3, and Fe5 spin-down (BS7-235) is energetically preferred. The electronic structure of the [VFeSC(CO)] BS7-235 model is compared to our [MoFeSC] BS7-235 model of FeMoco via localized orbital analysis and is discussed in terms of local oxidation states and different degrees of delocalization. As previously found from Fe X-ray absorption spectroscopy studies, the Fe part of FeVco is reduced compared to FeMoco, and the calculations reveal Fe5 as locally ferrous. This suggests resting-state FeVco to be analogous to an unprotonated E state of FeMoco. Furthermore, V-Fe interactions in FeVco are not as strong compared to Mo-Fe interactions in FeMoco. These clear differences in the electronic structures of otherwise similar cofactors suggest an explanation for distinct differences in reactivity.
固氮酶负责所有生物固氮过程。然而,这一过程在原子层面是如何实现的,目前仍未明确。依赖钼的固氮酶已得到广泛研究,是固氮酶中还原二氮最具活性的催化剂。另一方面,依赖钒的固氮酶表现出不同的反应活性,能够将一氧化碳和二氧化碳还原为碳氢化合物。直到最近,钒固氮酶的VFe蛋白晶体结构才得以确定,为在蛋白质基质中对铁钒辅因子(FeVco)进行详细的理论研究铺平了道路。晶体结构揭示了两个铁原子之间的一个桥连四原子配体,推测为一氧化碳或一氧化氮配体。利用VFe蛋白的量子力学/分子力学模型,从1.35 Å的晶体结构出发,我们系统地探索了FeVco的多种计算模型,考虑了一氧化碳或一氧化氮配体、三种不同的氧化还原状态以及多种破缺对称状态。我们发现,只有FeVco的[VFeSC(CO)]模型能很好地重现FeVco的晶体结构,这在计算模型中铁-铁和钒-铁距离的比较中可以看出。此外,具有Fe2、Fe3和Fe5自旋向下的破缺对称解(BS7-235)在能量上更占优势。通过局域轨道分析,将[VFeSC(CO)] BS7-235模型的电子结构与我们的FeMoco的[MoFeSC] BS7-235模型进行了比较,并从局部氧化态和不同程度的离域化角度进行了讨论。正如之前从铁X射线吸收光谱研究中发现的那样,与FeMoco相比,FeVco的铁部分被还原,计算结果显示Fe5为局部亚铁态。这表明静止状态的FeVco类似于FeMoco的未质子化E态。此外,与FeMoco中的钼-铁相互作用相比,FeVco中的钒-铁相互作用没有那么强。这些在其他方面相似的辅因子在电子结构上的明显差异为反应活性的显著差异提供了一种解释。