Bioinformatics, Department of Physics, Chemistry and Biology, Linköping University, Linköping, Sweden.
School of Bioscience, Systems Biology Research Center, University of Skövde, Skövde, Sweden.
NPJ Syst Biol Appl. 2022 Feb 23;8(1):9. doi: 10.1038/s41540-022-00218-9.
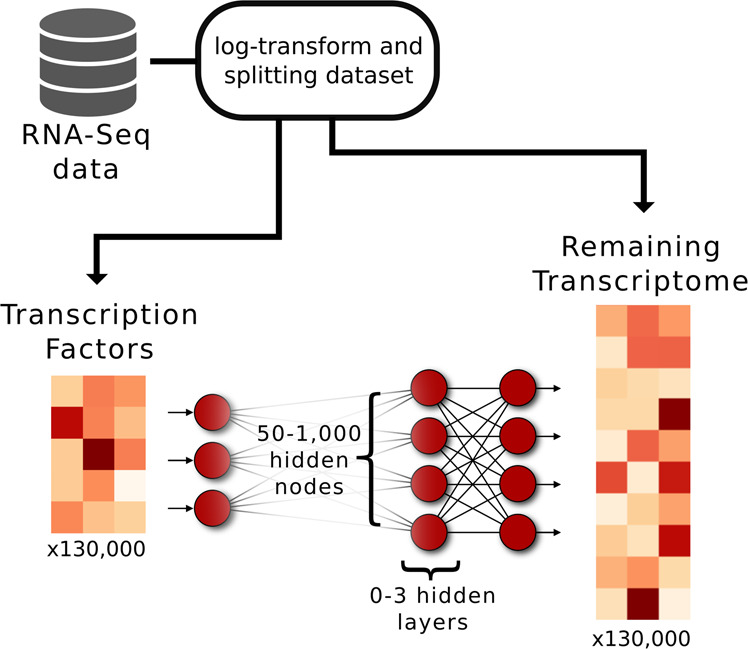
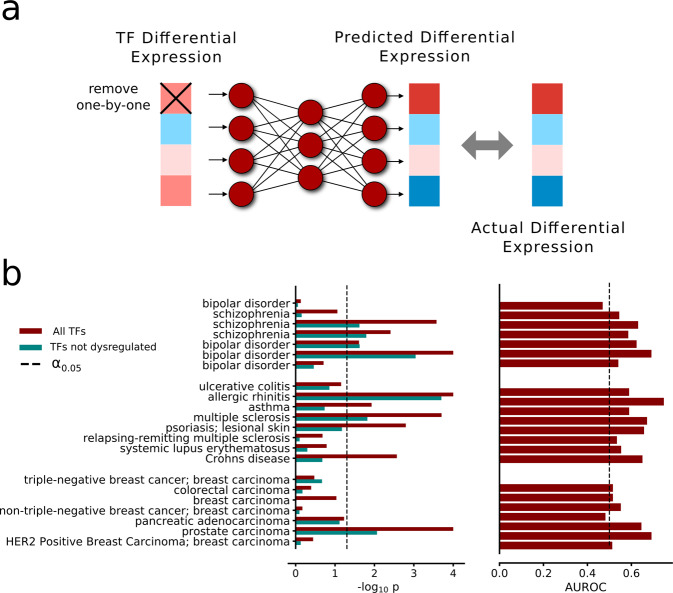
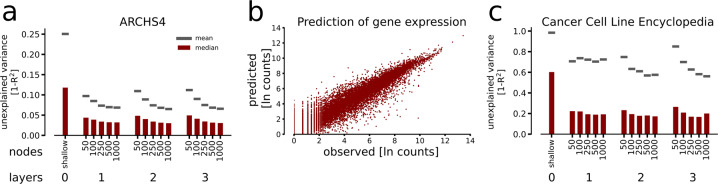
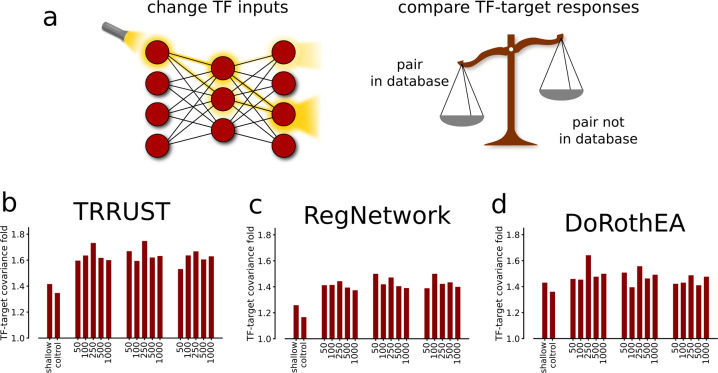
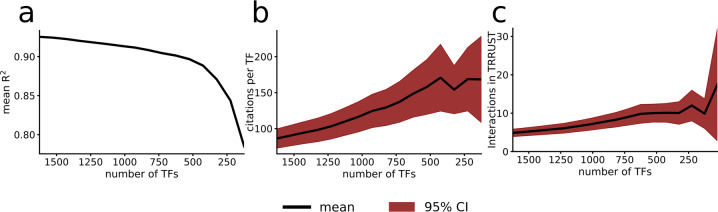
Prediction algorithms for protein or gene structures, including transcription factor binding from sequence information, have been transformative in understanding gene regulation. Here we ask whether human transcriptomic profiles can be predicted solely from the expression of transcription factors (TFs). We find that the expression of 1600 TFs can explain >95% of the variance in 25,000 genes. Using the light-up technique to inspect the trained NN, we find an over-representation of known TF-gene regulations. Furthermore, the learned prediction network has a hierarchical organization. A smaller set of around 125 core TFs could explain close to 80% of the variance. Interestingly, reducing the number of TFs below 500 induces a rapid decline in prediction performance. Next, we evaluated the prediction model using transcriptional data from 22 human diseases. The TFs were sufficient to predict the dysregulation of the target genes (rho = 0.61, P < 10). By inspecting the model, key causative TFs could be extracted for subsequent validation using disease-associated genetic variants. We demonstrate a methodology for constructing an interpretable neural network predictor, where analyses of the predictors identified key TFs that were inducing transcriptional changes during disease.
预测蛋白质或基因结构的算法,包括从序列信息预测转录因子结合,在理解基因调控方面具有变革性。在这里,我们想知道人类转录组谱是否可以仅从转录因子(TFs)的表达中预测。我们发现,1600 个 TF 的表达可以解释 25000 个基因中 >95%的方差。使用点亮技术检查训练好的神经网络,我们发现已知 TF-基因调控的过度表现。此外,学习到的预测网络具有层次化的组织。大约 125 个核心 TF 可以解释接近 80%的方差。有趣的是,将 TF 的数量减少到 500 以下会导致预测性能的快速下降。接下来,我们使用 22 种人类疾病的转录组数据评估了预测模型。TFs 足以预测靶基因的失调(rho=0.61,P<10)。通过检查模型,可以提取关键的致病 TF,以便使用与疾病相关的遗传变异进行后续验证。我们展示了一种构建可解释神经网络预测器的方法,其中对预测器的分析确定了在疾病期间诱导转录变化的关键 TF。