Quantitative Biosciences Institute (QBI), University of California San Francisco, San Francisco, California 94158, United States.
J. David Gladstone Institutes, San Francisco, California 94158, United States.
J Proteome Res. 2022 Apr 1;21(4):1124-1136. doi: 10.1021/acs.jproteome.1c00960. Epub 2022 Mar 2.
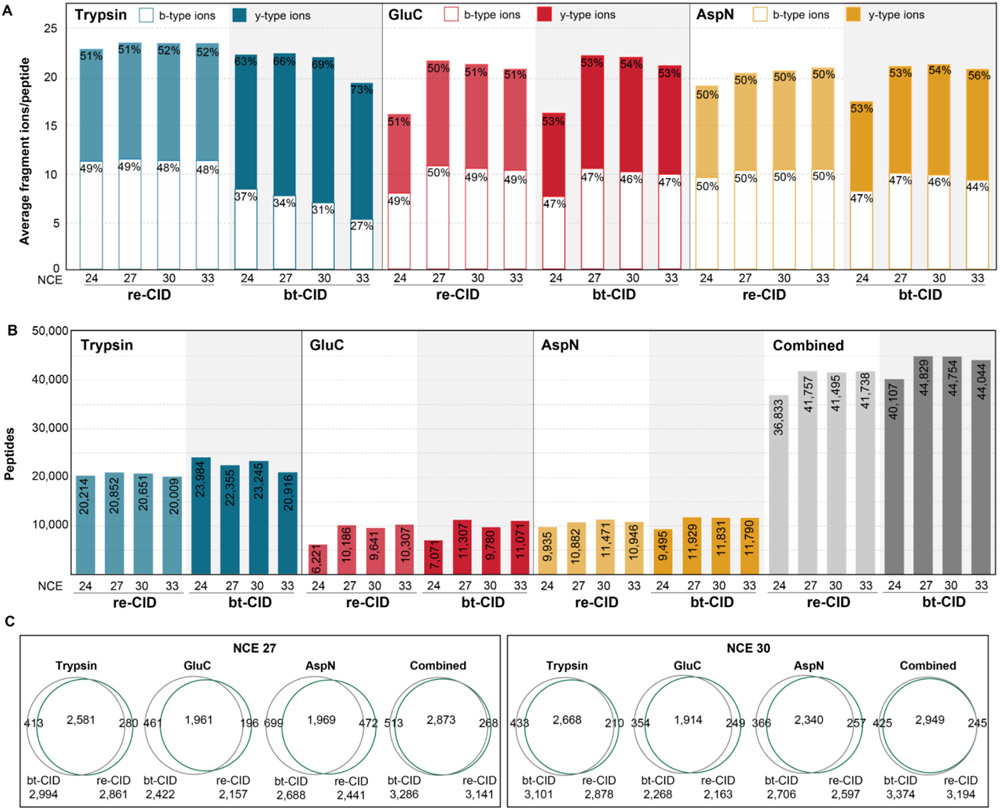
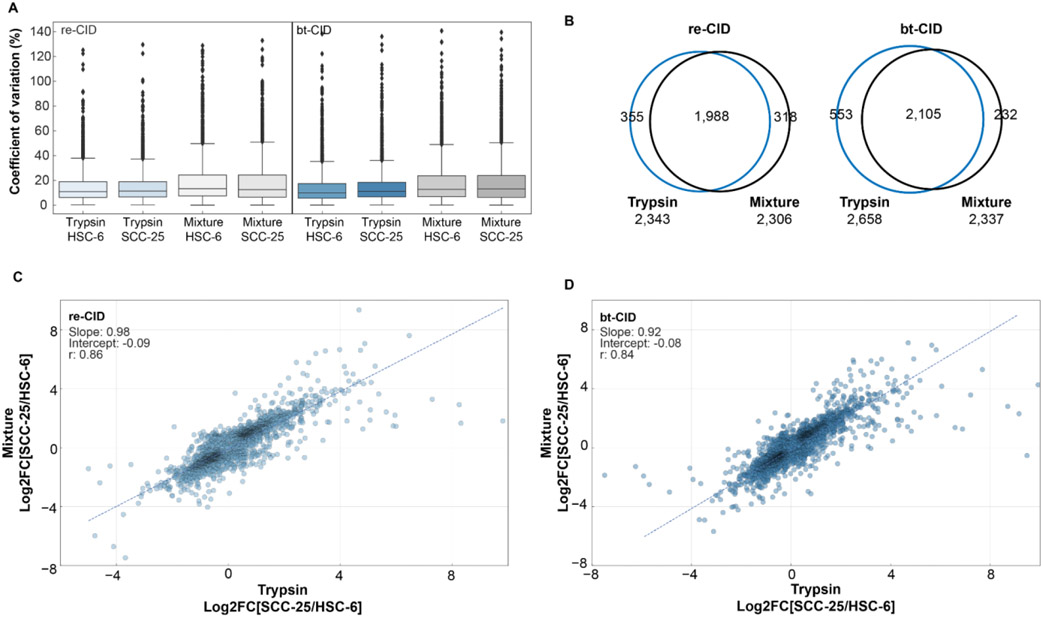
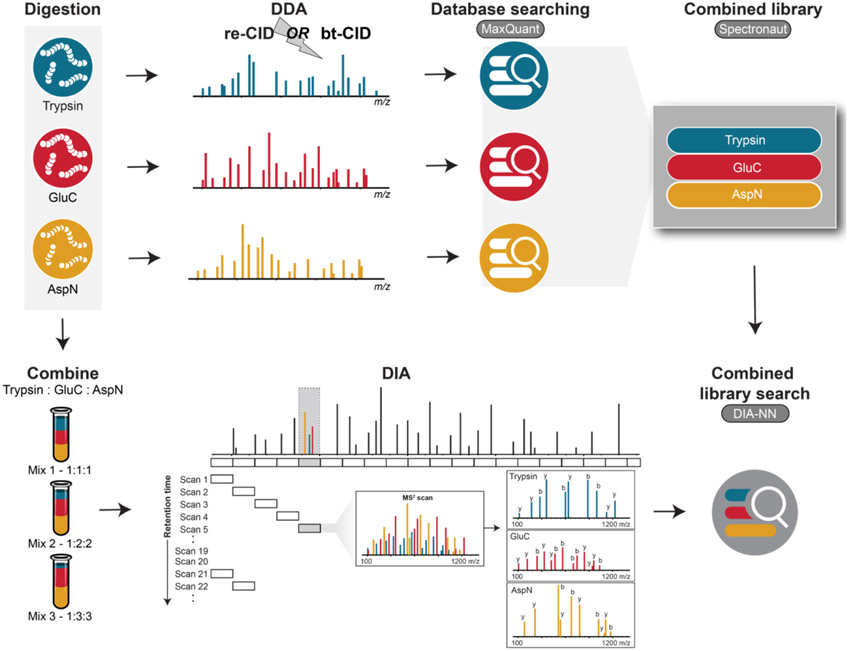
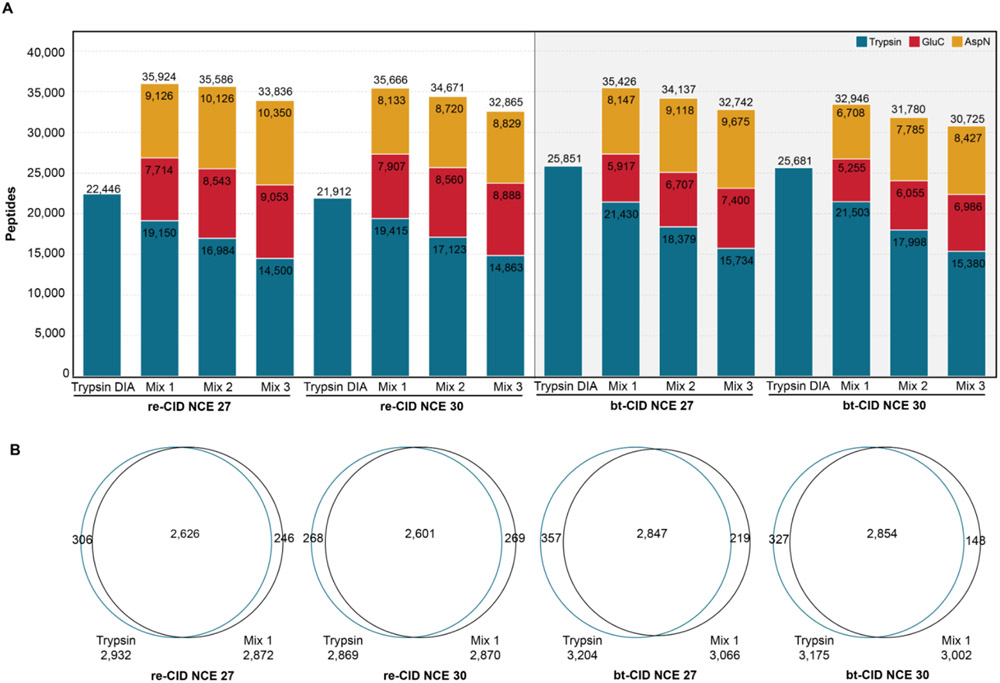
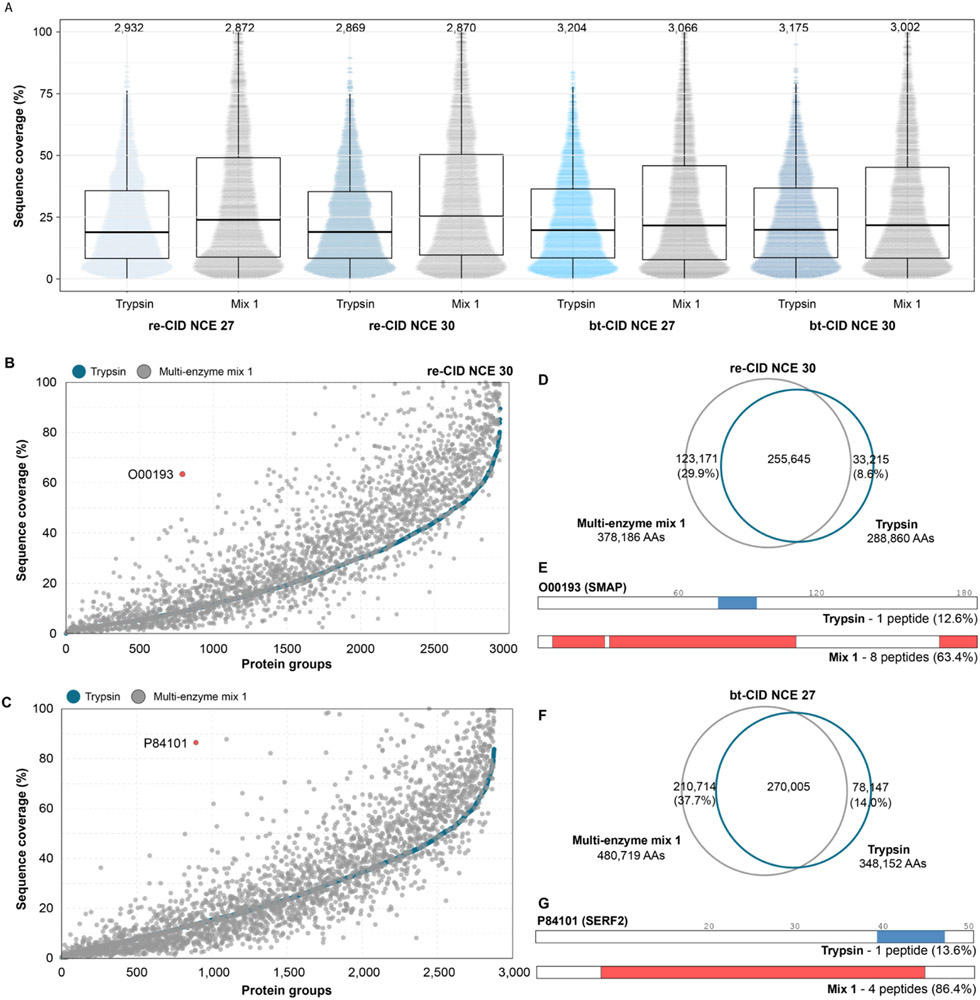
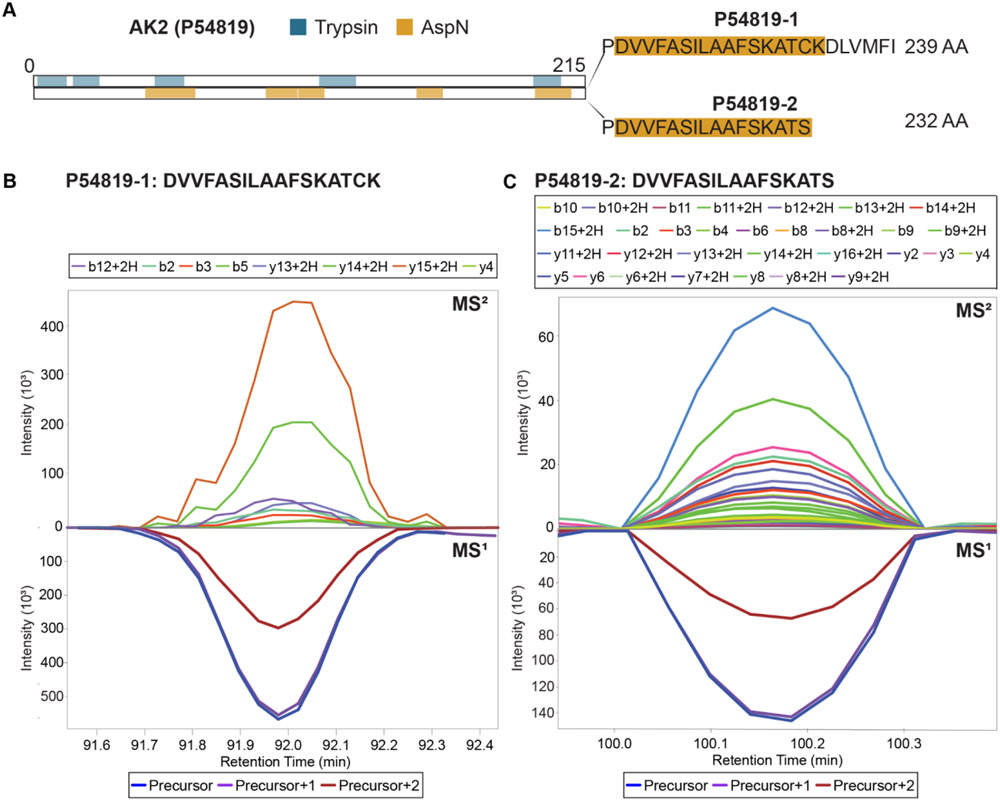
The use of multiple proteases has been shown to increase protein sequence coverage in proteomics experiments; however, due to the additional analysis time required, it has not been widely adopted in routine data-dependent acquisition (DDA) proteomic workflows. Alternatively, data-independent acquisition (DIA) has the potential to analyze multiplexed samples from different protease digests, but has been primarily optimized for fragmenting tryptic peptides. Here we evaluate a DIA multiplexing approach that combines three proteolytic digests (Trypsin, AspN, and GluC) into a single sample. We first optimize data acquisition conditions for each protease individually with both the canonical DIA fragmentation mode (beam type CID), as well as resonance excitation CID, to determine optimal consensus conditions across proteases. Next, we demonstrate that application of these conditions to a protease-multiplexed sample of human peptides results in similar protein identifications and quantitative performance as compared to trypsin alone, but enables up to a 63% increase in peptide detections, and a 45% increase in nonredundant amino acid detections. Nontryptic peptides enabled noncanonical protein isoform determination and resulted in 100% sequence coverage for numerous proteins, suggesting the utility of this approach in applications where sequence coverage is critical, such as protein isoform analysis.
使用多种蛋白酶已被证明可以增加蛋白质组学实验中的蛋白质序列覆盖度;然而,由于需要额外的分析时间,它并未在常规的数据依赖型采集(DDA)蛋白质组学工作流程中广泛采用。相比之下,非依赖性数据采集(DIA)有可能分析来自不同蛋白酶消化物的多路复用样品,但主要针对胰蛋白酶肽的片段化进行了优化。在这里,我们评估了一种 DIA 多路复用方法,该方法将三种酶切(胰蛋白酶、AspN 和 GluC)组合到一个样品中。我们首先使用经典的 DIA 碎裂模式(束型 CID)以及共振激发 CID 分别优化每种蛋白酶的采集条件,以确定跨蛋白酶的最佳共识条件。接下来,我们证明将这些条件应用于人类肽的蛋白酶多路复用样品中,与单独使用胰蛋白酶相比,在蛋白质鉴定和定量性能方面相似,但可将肽检测的数量增加多达 63%,非冗余氨基酸检测的数量增加 45%。非胰蛋白酶肽可实现非典型蛋白质同工型的测定,并对许多蛋白质实现了 100%的序列覆盖,这表明该方法在需要序列覆盖度的应用中具有实用性,例如蛋白质同工型分析。