Narayan Reema, Gadag Shivaprasad, Garg Sanjay, Nayak Usha Y
Department of Pharmaceutics, Manipal College of Pharmaceutical Sciences, Manipal Academy of Higher Education, Manipal 576104 Karnataka, India.
UniSA: Clinical and Health Sciences, University of South Australia, Adelaide, South Australia 5000, Australia.
ACS Omega. 2022 Mar 4;7(10):8229-8245. doi: 10.1021/acsomega.1c03618. eCollection 2022 Mar 15.
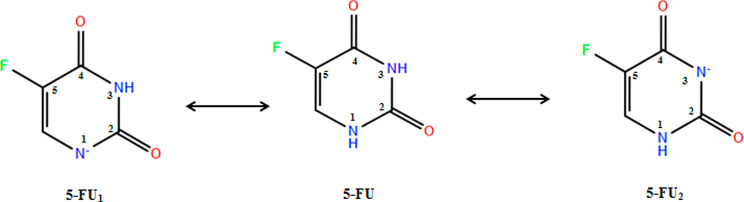
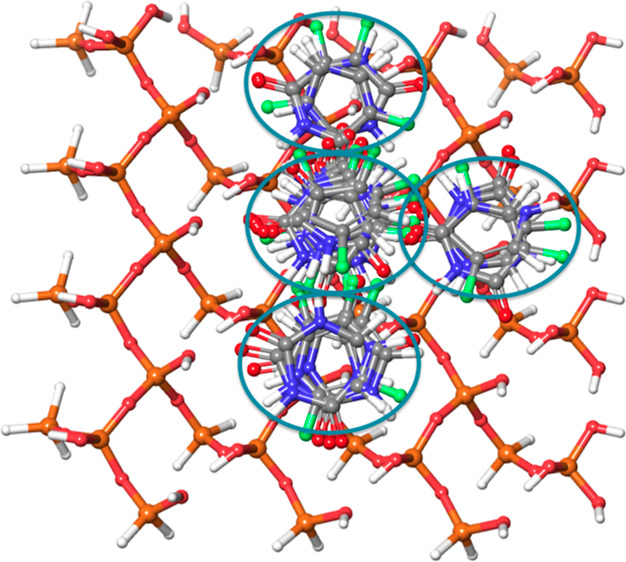
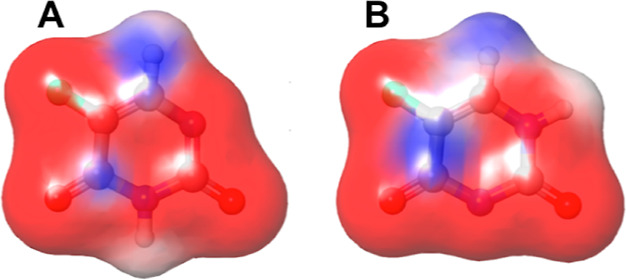
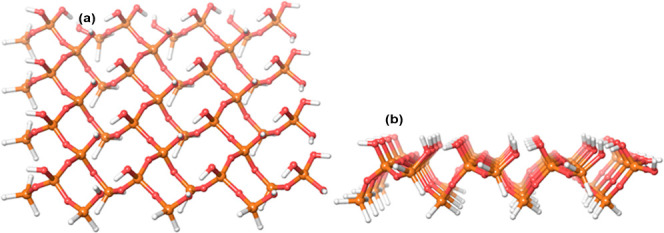
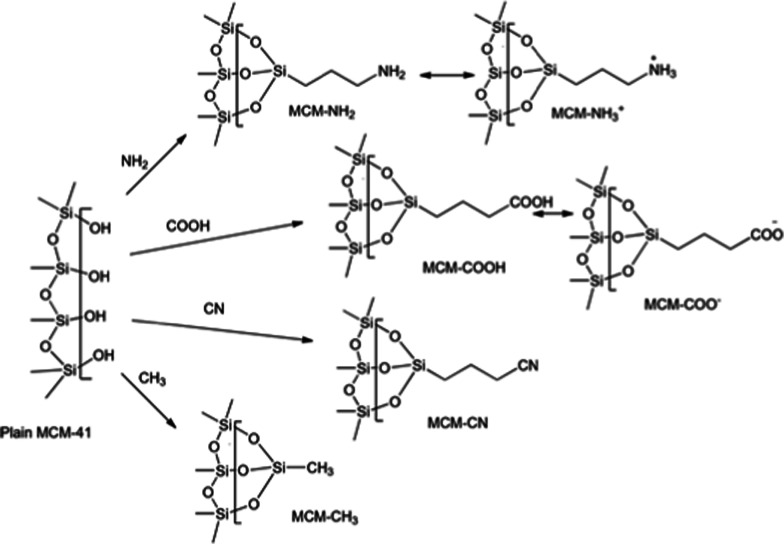
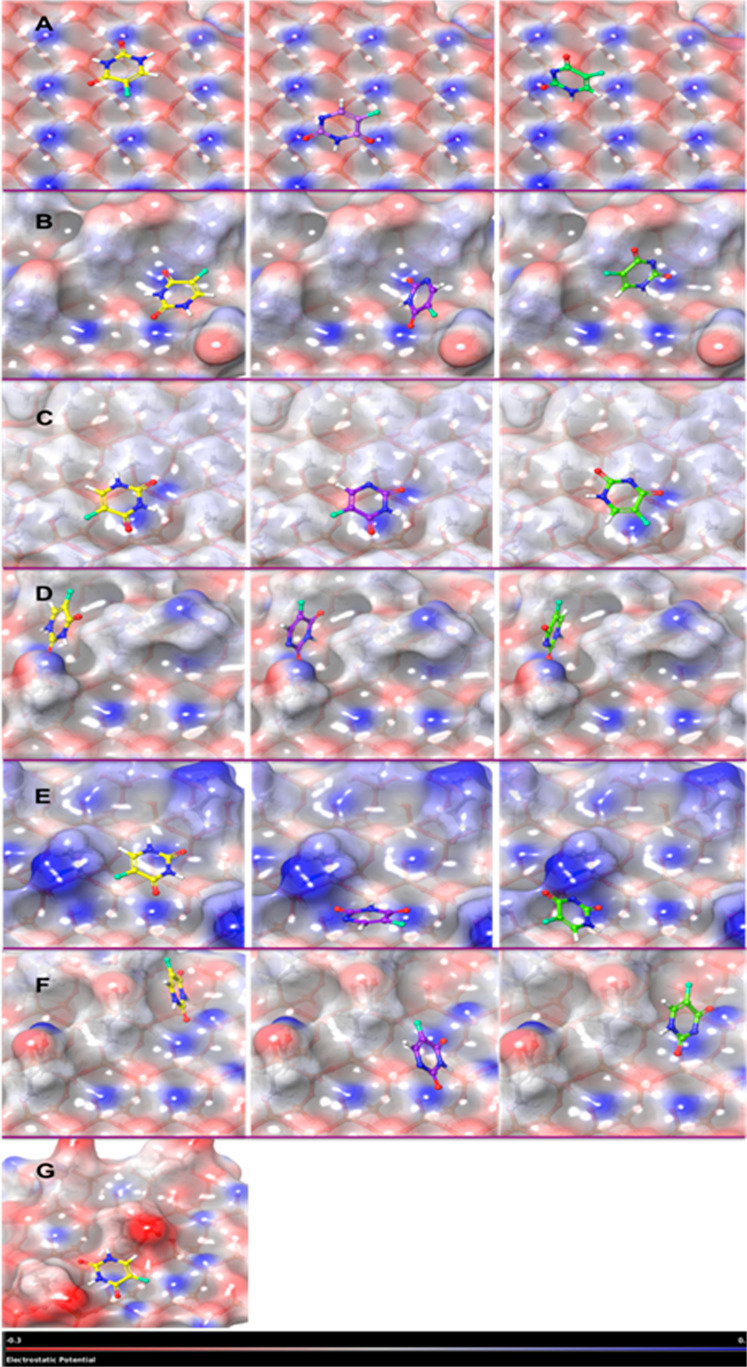
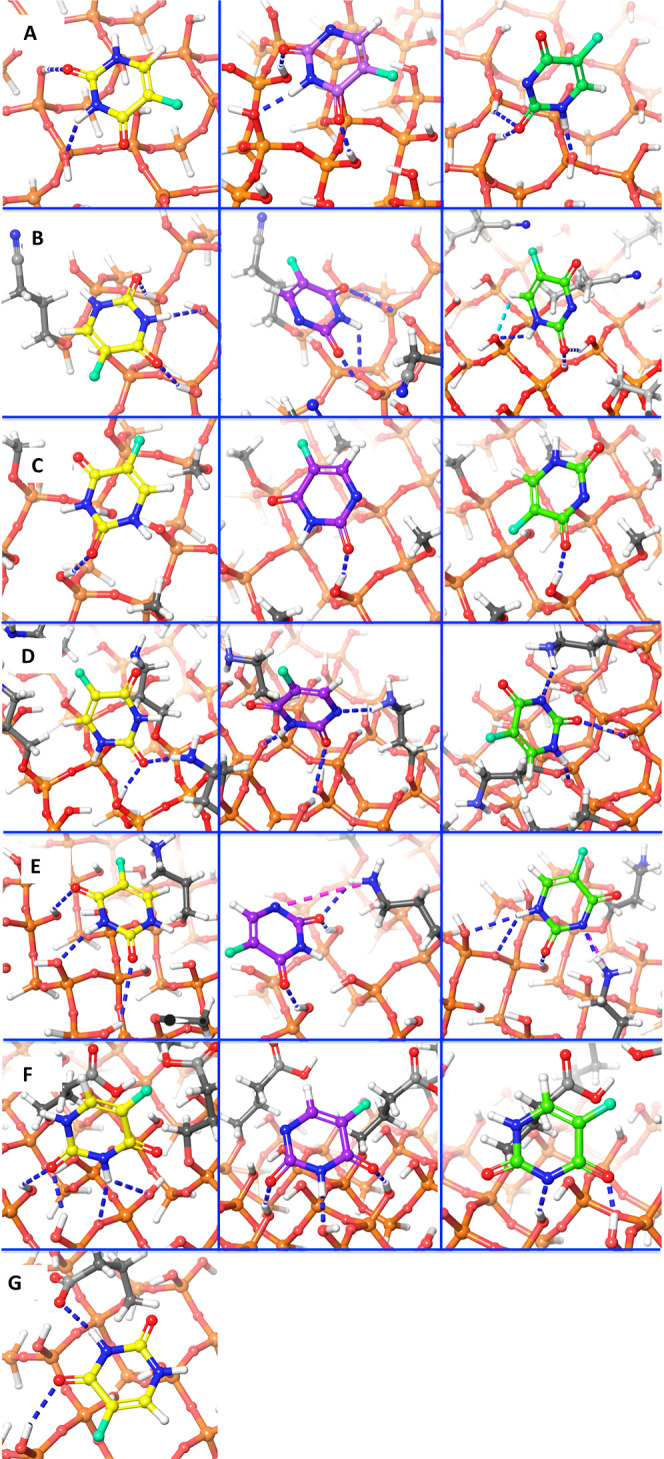
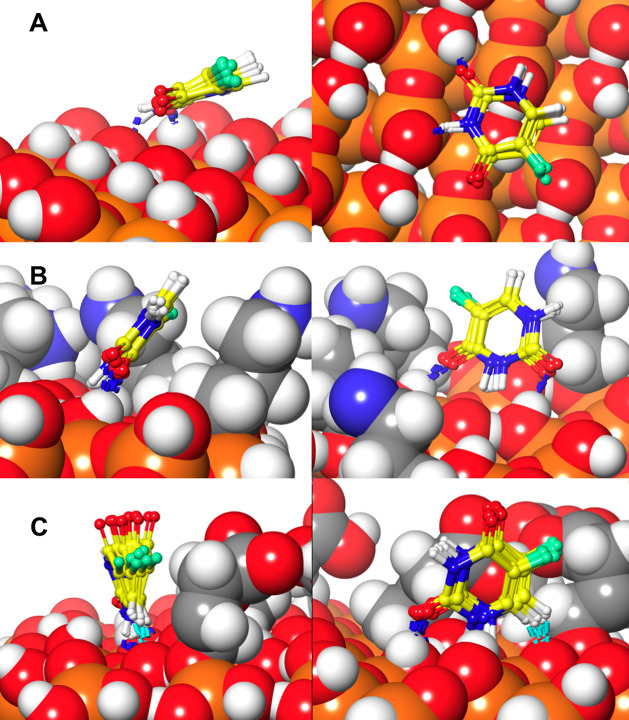
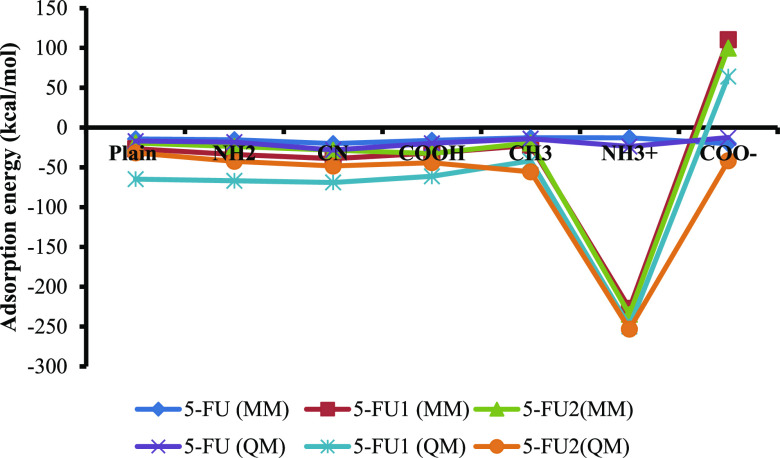
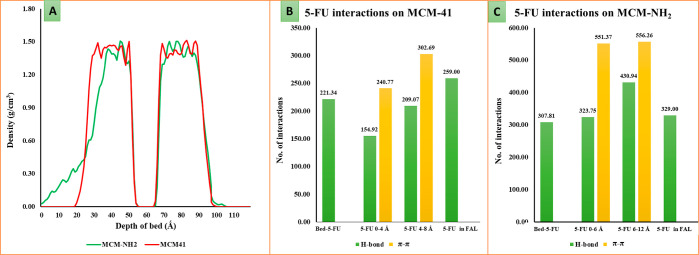
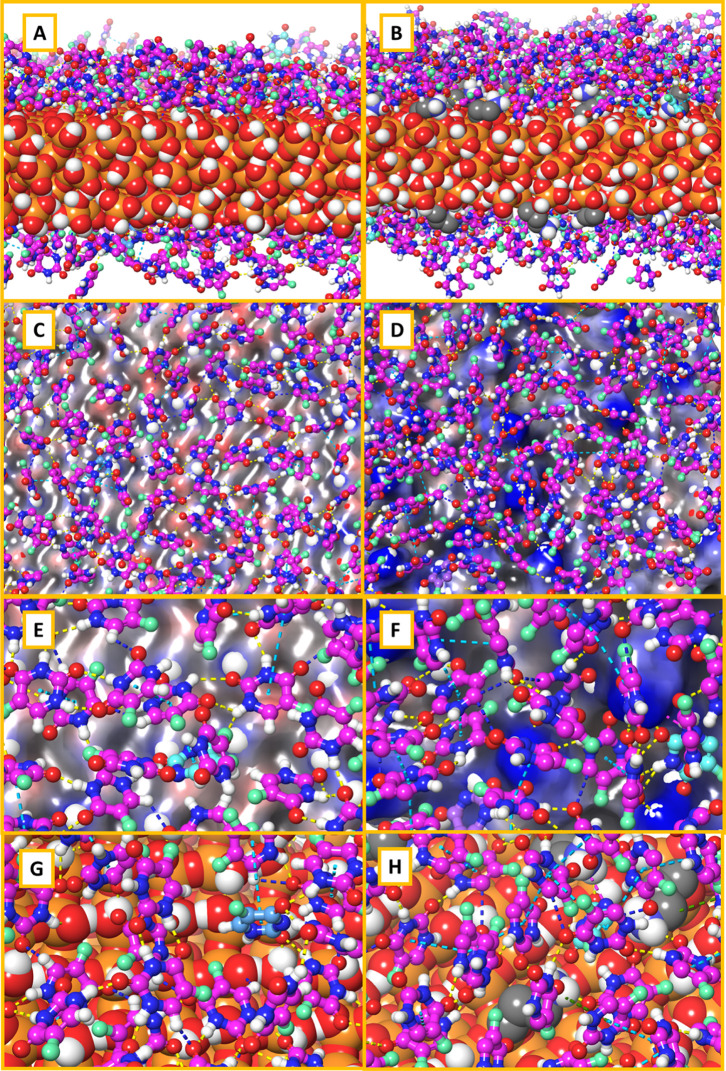
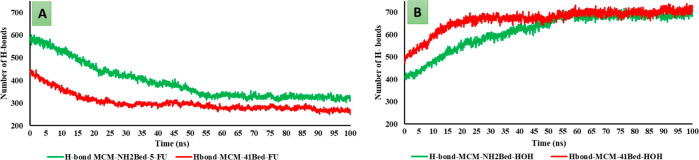
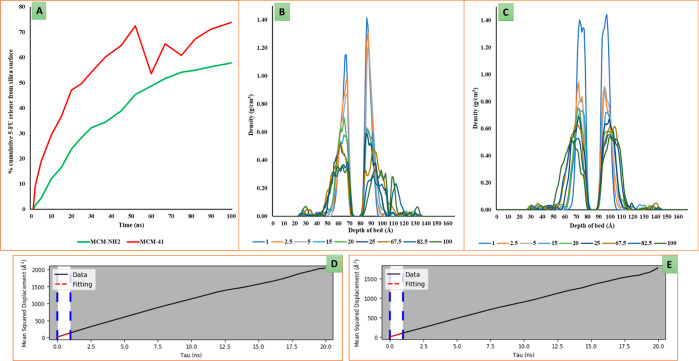
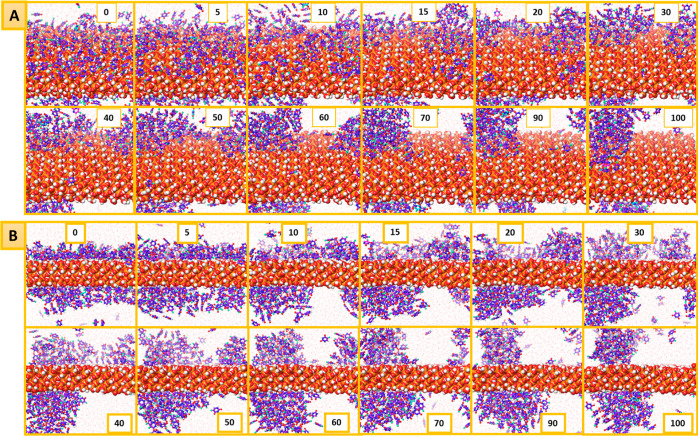
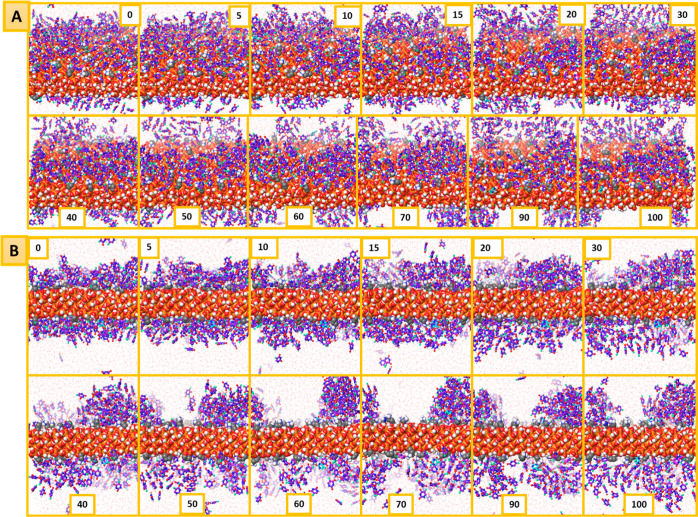
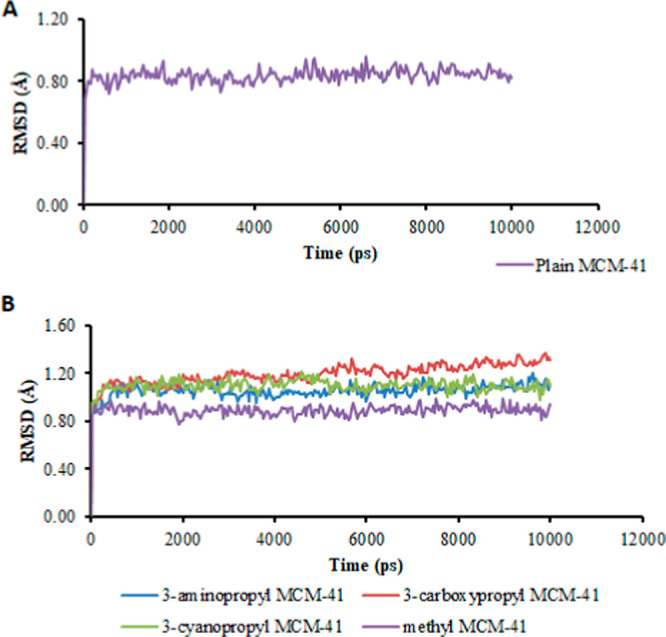
MCM-41, a type of mesoporous silica nanoparticle, has garnered widespread interests as a useful carrier for drug delivery wherein the drug gets adsorbed into the pores of the carrier. To understand the adsorption mechanism and release of the drug at the molecular level, in the current study, it was attempted to generate a computational model for the loading of 5-fluorouracil (5-FU), a chemotherapeutic agent into surface-modified MCM-41. The molecular surface models of the mesoporous silica (MCM-41) nanoparticle with different surface substitutions were created. In the first stage, molecular mechanics (MM) simulations were carried out to obtain the optimized surface structures. Subsequently, a 5-FU drug molecule in its different forms was docked on top of different MCM-41 surfaces to understand the adsorption orientation and energetics. To further validate the results, more accurate quantum mechanical (QM) calculations were also carried out, and the energetics between the QM and MM calculations are found to be similar. All the substitutions (-NH, -CN, -COOH) except the methyl substitution exhibited favorable interactions compared to the unsubstituted MCM-41 surface which was in accordance with the experimental observations. The release rate of 5-FU from MCM-41 and aminopropyl-substituted MCM-41 (MCM-NH) was studied using molecular dynamics simulations which revealed that the release rate of 5-FU from the MCM-NH surface was slower compared to that of plain MCM-41. The detailed surface characteristics and the adsorption energies from the molecular simulations correlating the loading capacity and release are reported in here.
MCM - 41是一种介孔二氧化硅纳米颗粒,作为一种有用的药物递送载体已引起广泛关注,药物可吸附在载体的孔中。为了在分子水平上理解药物的吸附机制和释放过程,在当前研究中,尝试建立一个将化疗药物5 - 氟尿嘧啶(5 - FU)负载到表面改性的MCM - 41中的计算模型。创建了具有不同表面取代的介孔二氧化硅(MCM - 41)纳米颗粒的分子表面模型。在第一阶段,进行分子力学(MM)模拟以获得优化的表面结构。随后,将不同形式的5 - FU药物分子对接在不同的MCM - 41表面上,以了解吸附方向和能量学。为了进一步验证结果,还进行了更精确的量子力学(QM)计算,发现QM和MM计算之间的能量学相似。与未取代的MCM - 41表面相比,除甲基取代外的所有取代(-NH、-CN、-COOH)均表现出有利的相互作用,这与实验观察结果一致。使用分子动力学模拟研究了5 - FU从MCM - 41和氨丙基取代的MCM - 41(MCM - NH)中的释放速率,结果表明5 - FU从MCM - NH表面的释放速率比普通MCM - 41慢。本文报道了详细的表面特征以及与负载能力和释放相关的分子模拟吸附能。