El Khoury Léa, Jing Zhifeng, Cuzzolin Alberto, Deplano Alessandro, Loco Daniele, Sattarov Boris, Hédin Florent, Wendeborn Sebastian, Ho Chris, El Ahdab Dina, Jaffrelot Inizan Theo, Sturlese Mattia, Sosic Alice, Volpiana Martina, Lugato Angela, Barone Marco, Gatto Barbara, Macchia Maria Ludovica, Bellanda Massimo, Battistutta Roberto, Salata Cristiano, Kondratov Ivan, Iminov Rustam, Khairulin Andrii, Mykhalonok Yaroslav, Pochepko Anton, Chashka-Ratushnyi Volodymyr, Kos Iaroslava, Moro Stefano, Montes Matthieu, Ren Pengyu, Ponder Jay W, Lagardère Louis, Piquemal Jean-Philip, Sabbadin Davide
Qubit Pharmaceuticals, Incubateur Paris Biotech Santé 24 Rue du Faubourg Saint Jacques 75014 Paris France
Chiesi Farmaceutici S.p.A, Nuovo Centro Ricerche Largo Belloli 11a 43122 Parma Italy.
Chem Sci. 2022 Feb 10;13(13):3674-3687. doi: 10.1039/d1sc05892d. eCollection 2022 Mar 30.
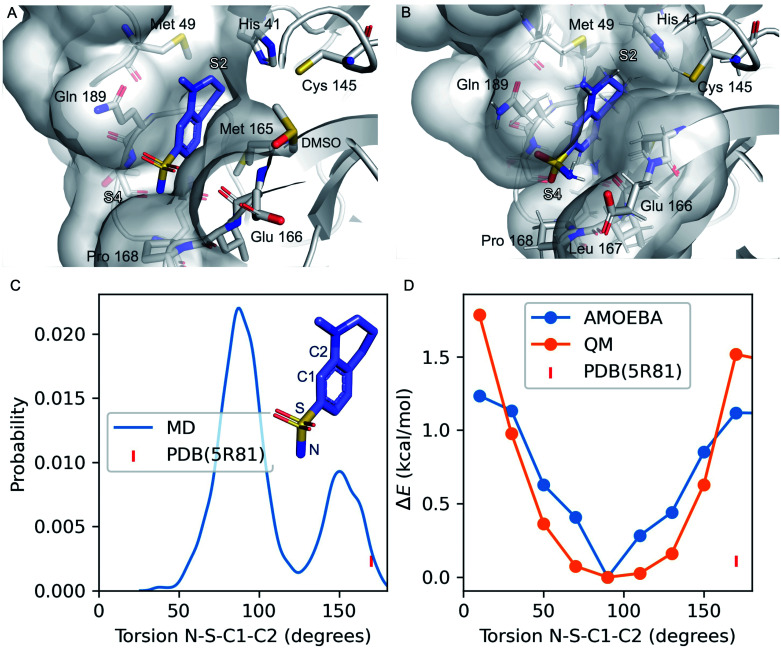
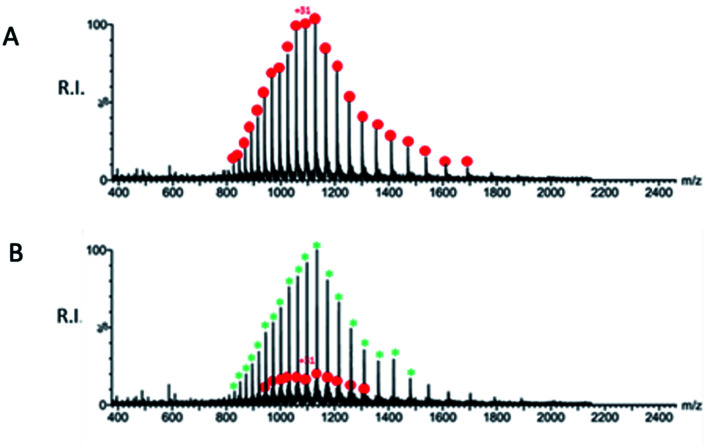
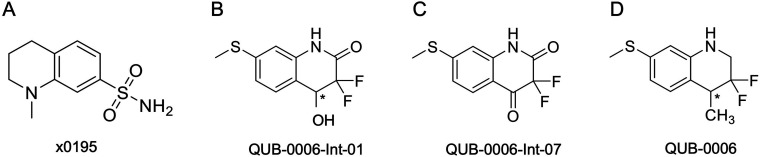
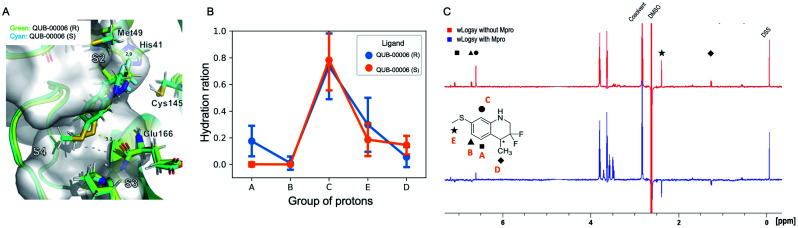
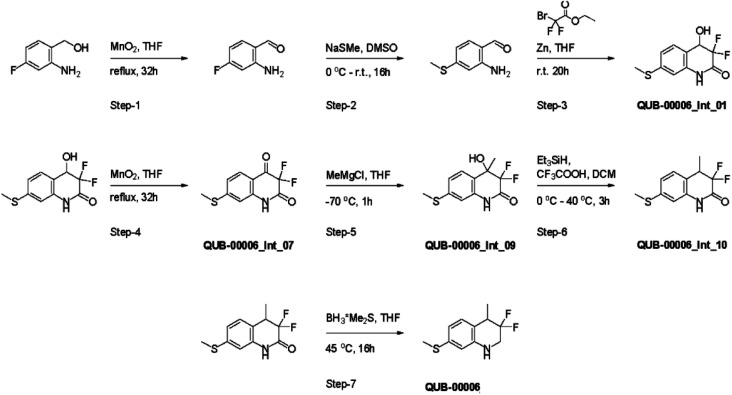
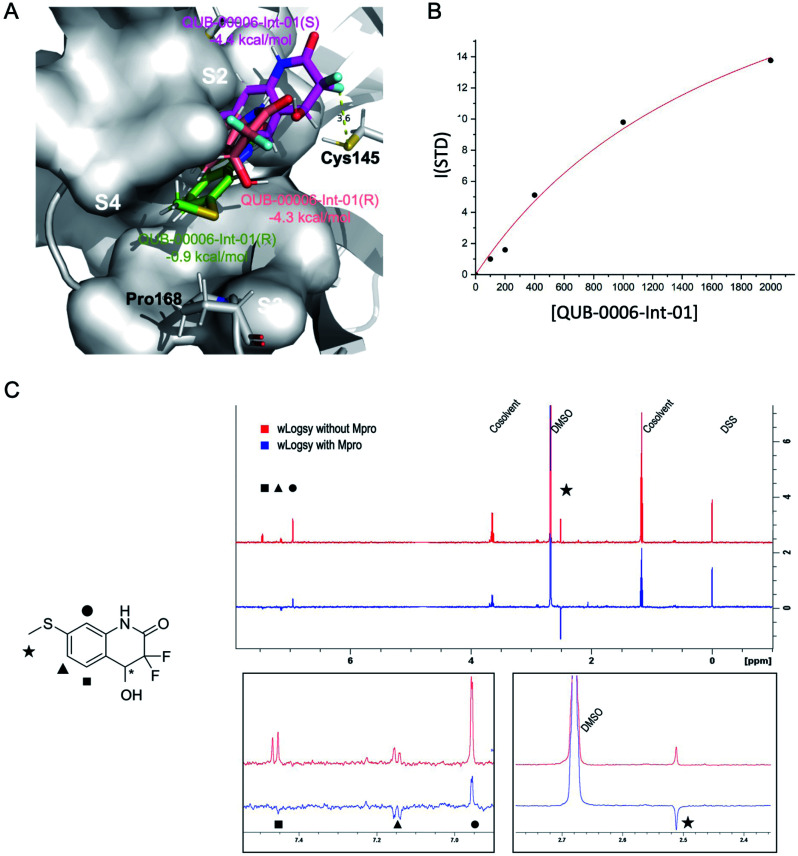
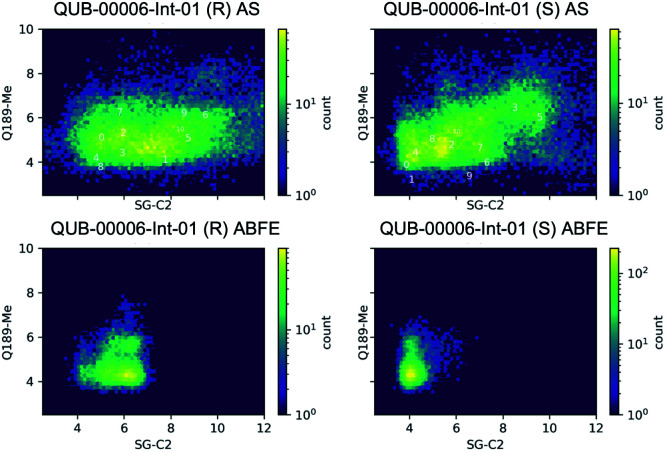
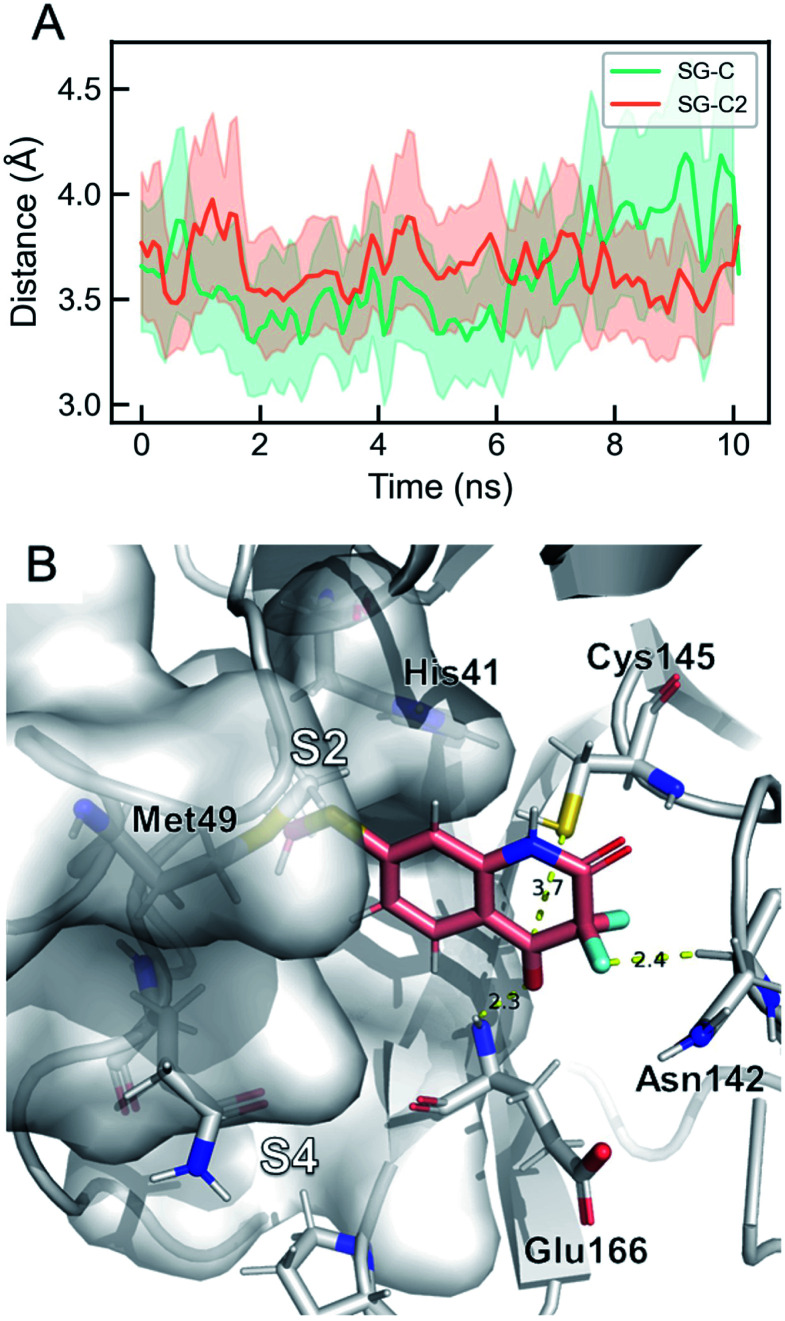
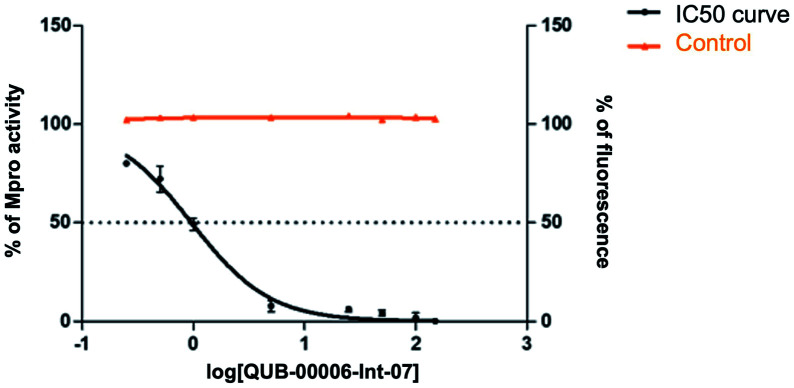
We report a fast-track computationally driven discovery of new SARS-CoV-2 main protease (M) inhibitors whose potency ranges from mM for the initial non-covalent ligands to sub-μM for the final covalent compound (IC = 830 ± 50 nM). The project extensively relied on high-resolution all-atom molecular dynamics simulations and absolute binding free energy calculations performed using the polarizable AMOEBA force field. The study is complemented by extensive adaptive sampling simulations that are used to rationalize the different ligand binding poses through the explicit reconstruction of the ligand-protein conformation space. Machine learning predictions are also performed to predict selected compound properties. While simulations extensively use high performance computing to strongly reduce the time-to-solution, they were systematically coupled to nuclear magnetic resonance experiments to drive synthesis and for characterization of compounds. Such a study highlights the power of strategies that rely on structure-based approaches for drug design and allows the protein conformational multiplicity problem to be addressed. The proposed fluorinated tetrahydroquinolines open routes for further optimization of M inhibitors towards low nM affinities.
我们报告了一种通过计算驱动的快速发现新型严重急性呼吸综合征冠状病毒2(SARS-CoV-2)主要蛋白酶(M)抑制剂的方法,其效力范围从最初非共价配体的毫摩尔级到最终共价化合物的亚微摩尔级(IC = 830 ± 50 nM)。该项目广泛依赖于使用可极化的AMOEBA力场进行的高分辨率全原子分子动力学模拟和绝对结合自由能计算。通过广泛的自适应采样模拟对该研究进行补充,这些模拟用于通过明确重建配体 - 蛋白质构象空间来合理化不同的配体结合姿势。还进行了机器学习预测以预测选定的化合物性质。虽然模拟广泛使用高性能计算以大幅缩短解决问题的时间,但它们系统地与核磁共振实验相结合,以推动化合物的合成和表征。这样的研究突出了依赖基于结构的药物设计方法的策略的力量,并允许解决蛋白质构象多样性问题。所提出的氟化四氢喹啉为进一步优化M抑制剂以实现低纳摩尔亲和力开辟了途径。