KwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), School of Laboratory Medicine & Medical Sciences, University of KwaZulu-Natal Durban 4001, KwaZulu-Natal, South Africa.
Centre for Epidemic Response and Innovation (CERI), Stellenbosch University, Stellenbosch, South Africa.
BMC Genomics. 2022 Apr 22;23(1):319. doi: 10.1186/s12864-022-08541-5.
Over 4 million SARS-CoV-2 genomes have been sequenced globally in the past 2 years. This has been crucial in elucidating transmission chains within communities, the development of new diagnostic methods, vaccines, and antivirals. Although several sequencing technologies have been employed, Illumina and Oxford Nanopore remain the two most commonly used platforms. The sequence quality between these two platforms warrants a comparison of the genomes produced by the two technologies. Here, we compared the SARS-CoV-2 consensus genomes obtained from the Oxford Nanopore Technology GridION and the Illumina MiSeq for 28 sequencing runs.
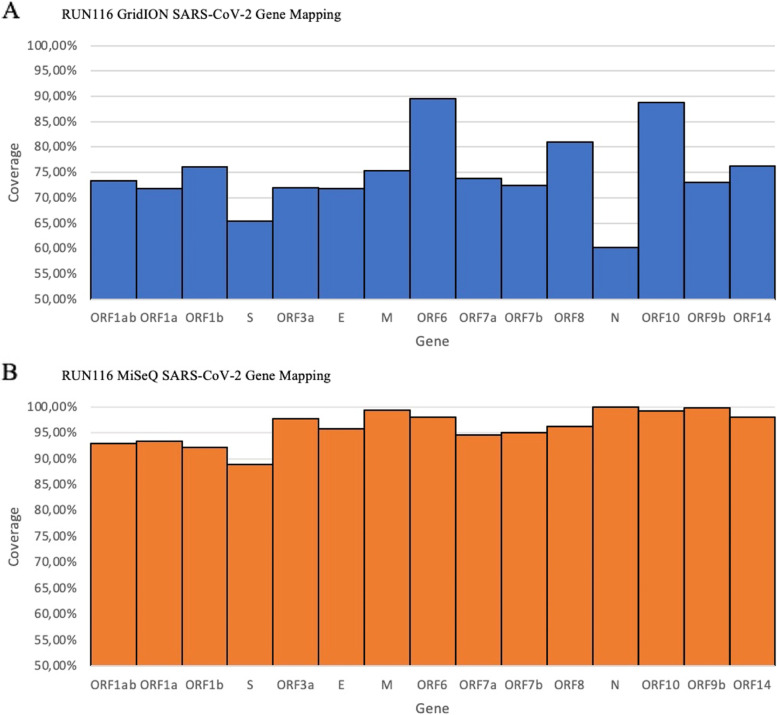
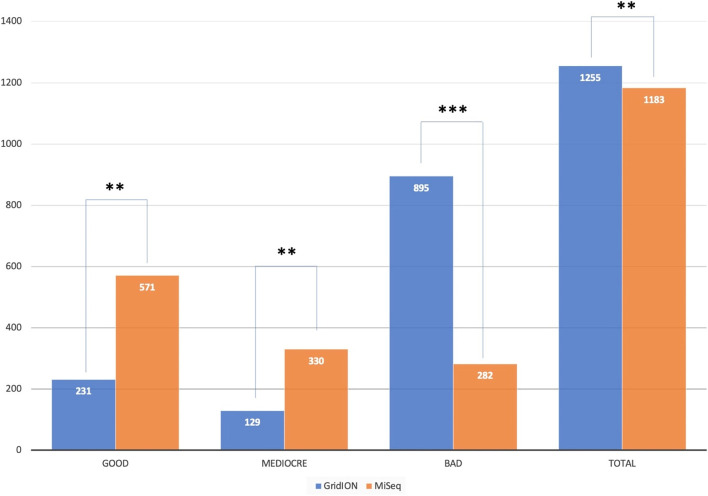
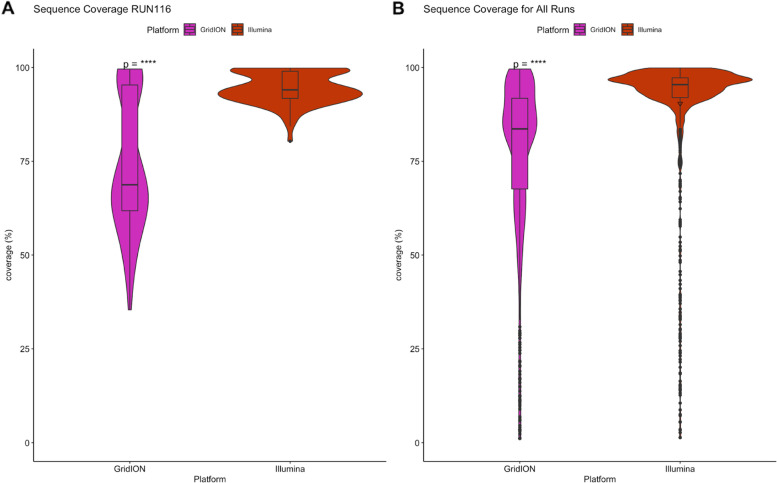
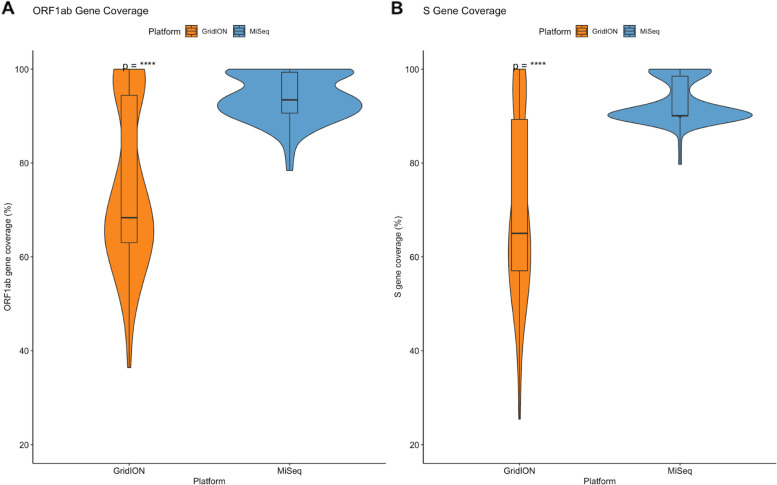
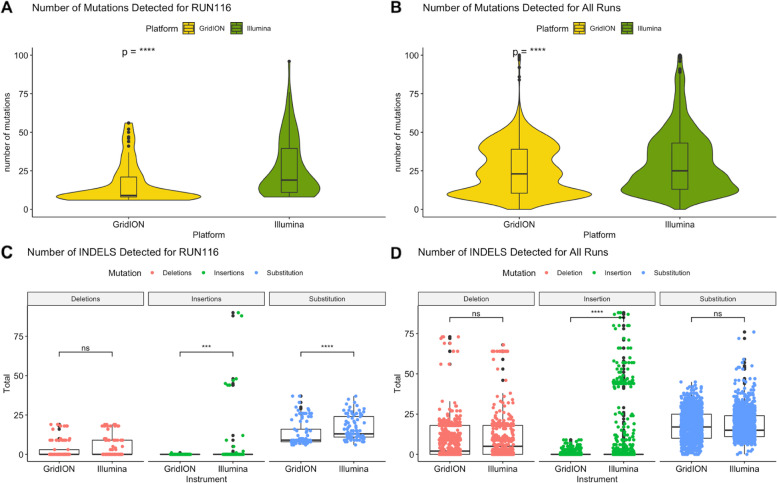
Our results show that the MiSeq had a significantly higher number of consensus genomes classified by Nextclade as good and mediocre compared to the GridION. The MiSeq also had a significantly higher genome coverage and mutation counts than the GridION.
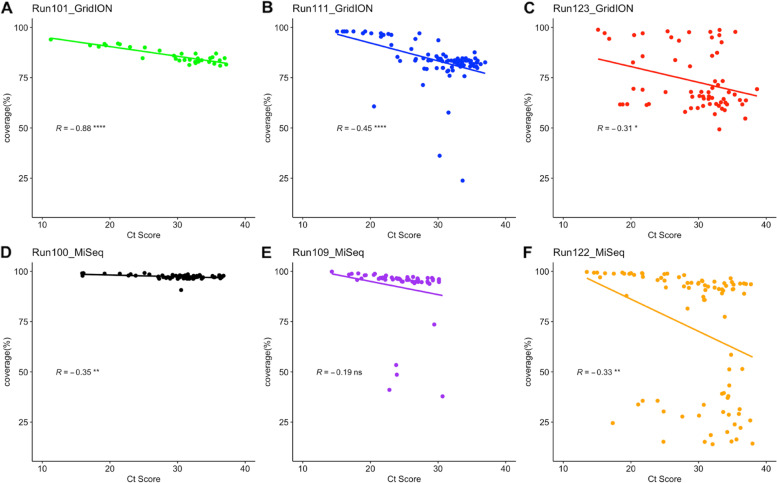
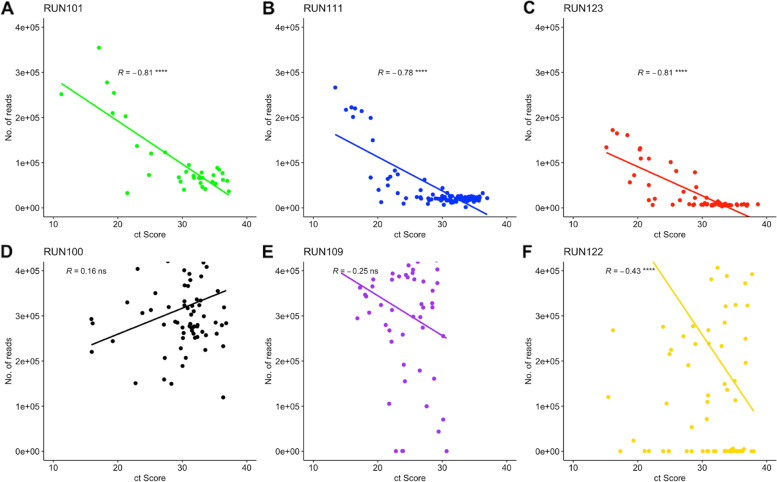
Due to the low genome coverage, high number of indels, and sensitivity to SARS-CoV-2 viral load noted with the GridION when compared to MiSeq, we can conclude that the MiSeq is more favourable for SARS-CoV-2 genomic surveillance, as successful genomic surveillance is dependent on high quality, near-whole consensus genomes.
在过去的 2 年中,已经在全球范围内对超过 400 万份 SARS-CoV-2 基因组进行了测序。这对于阐明社区内的传播链、开发新的诊断方法、疫苗和抗病毒药物至关重要。尽管已经采用了几种测序技术,但 Illumina 和 Oxford Nanopore 仍然是使用最广泛的两种平台。这两种平台的序列质量需要对两种技术产生的基因组进行比较。在这里,我们比较了来自 Oxford Nanopore Technology GridION 和 Illumina MiSeq 的 28 次测序运行的 SARS-CoV-2 共识基因组。
我们的结果表明,与 GridION 相比,Nextclade 将 MiSeq 分类为良好和中等的共识基因组数量明显更高。MiSeq 的基因组覆盖率和突变计数也明显高于 GridION。
与 MiSeq 相比,GridION 注意到低基因组覆盖率、高插入缺失(indels)数量以及对 SARS-CoV-2 病毒载量的敏感性,因此我们可以得出结论,MiSeq 更适合 SARS-CoV-2 基因组监测,因为成功的基因组监测依赖于高质量、近乎完整的共识基因组。