Department of Molecular Biosciences, University of Texas at Austin, Austin, Texas, USA.
mBio. 2022 Jun 28;13(3):e0058822. doi: 10.1128/mbio.00588-22. Epub 2022 May 2.
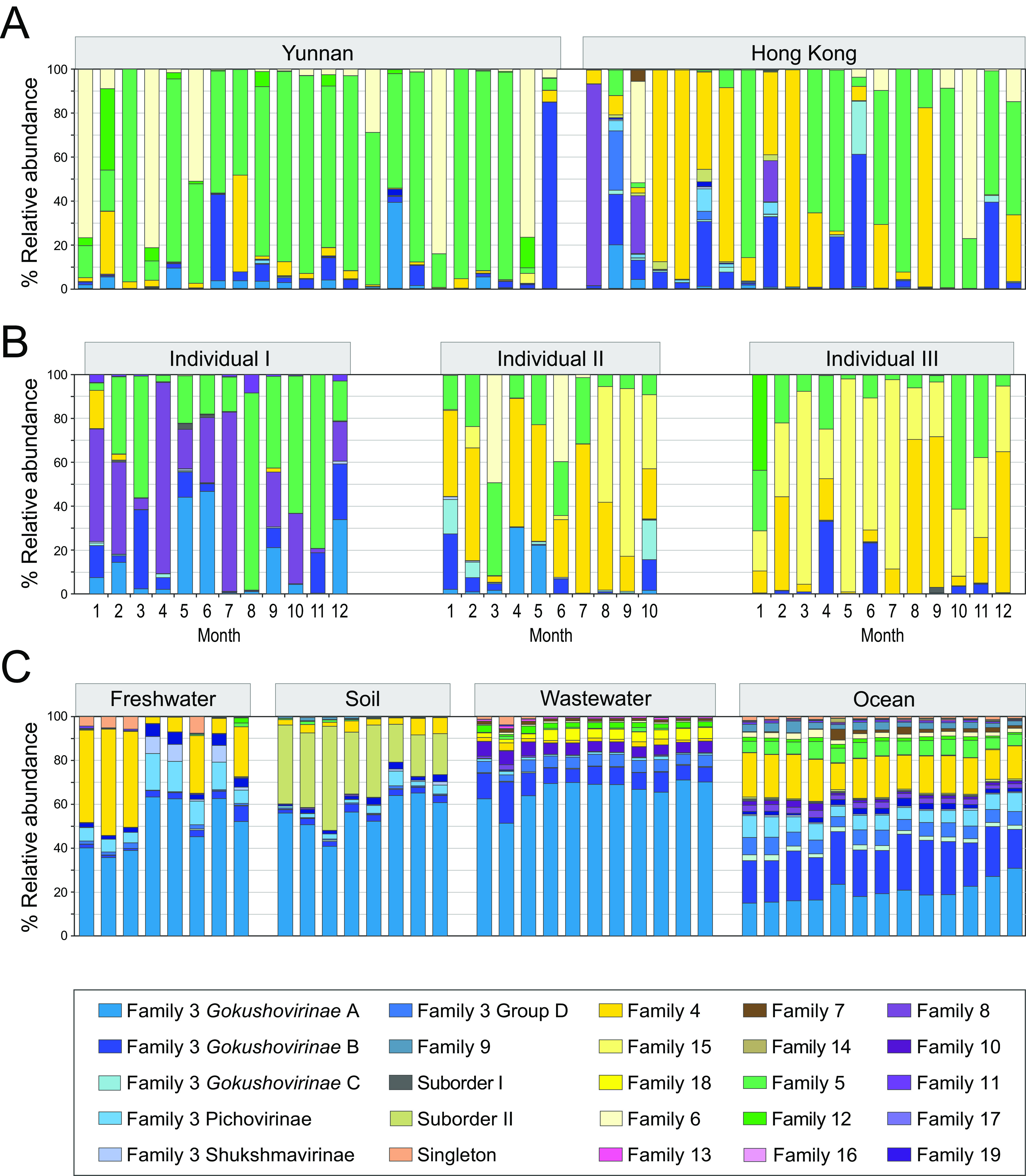
Microviruses encompass an astonishing array of small, single-stranded DNA phages that, due to the surge in metagenomic surveys, are now known to be prevalent in most environments. Current taxonomy concedes the considerable diversity within this lineage to a single family (the ), which has rendered it difficult to adequately and accurately assess the amount of variation that actually exists within this group. We amassed and curated the largest collection of microviral genomes to date and, through a combination of protein-sharing networks and phylogenetic analysis, discovered at least three meaningful taxonomic levels between the current ranks of family and genus. When considering more than 13,000 microviral genomes from recognized lineages and as-yet-unclassified microviruses in metagenomic samples, microviral diversity is better understood by elevating microviruses to the level of an order that consists of three suborders and at least 19 putative families, each with their respective subfamilies. These revisions enable fine-scale assessment of microviral dynamics: for example, in the human gut, there are considerable differences in the abundances of microviral families both between urban and rural populations and in individuals over time. In addition, our analysis of genome contents and gene exchange shows that microviral families carry no recognizable accessory metabolic genes and rarely, if ever, engage in horizontal gene transfer across microviral families or with their bacterial hosts. These insights bring microviral taxonomy in line with current developments in the taxonomy of other phages and increase the understanding of microvirus biology. Microviruses are the most abundant single-stranded DNA phages on the planet and an important component of the human gut virome. And yet, productive research into their biology is hampered by the inadequacies of current taxonomic ordering: microviruses are lumped into a single family and treated as a monolithic group, thereby obscuring the extent of their diversity and resulting in little comparative research. Our investigations into the diversity of microviruses define numerous groups, most lacking any isolated representatives, and point toward high-value targets for future research. To expedite microvirus discovery and comparison, we developed a pipeline that enables the fast and facile sorting of novel microvirus genomes into well-defined taxonomic groups. These improvements provide new insights into the biology of microviruses and emphasize fundamental differences between these miniature phages and their large, double-stranded DNA phage competitors.
微病毒涵盖了令人惊讶的一系列小型单链 DNA 噬菌体,由于宏基因组调查的激增,现在已知它们在大多数环境中普遍存在。目前的分类学承认该谱系内存在相当大的多样性,仅归属于一个科(),这使得难以充分和准确地评估该群体中实际存在的变异量。我们收集并整理了迄今为止最大的微病毒基因组集合,并通过蛋白质共享网络和系统发育分析的组合,发现了当前科和属等级之间至少存在三个有意义的分类学水平。当考虑来自已识别谱系和宏基因组样本中尚未分类的微病毒的超过 13000 个微病毒基因组时,通过将微病毒提升到由三个亚目和至少 19 个假定科组成的一个目,可以更好地理解微病毒的多样性,每个科都有各自的亚科。这些修订使微病毒的动态能够进行精细评估:例如,在人类肠道中,微病毒家族的丰度在城市和农村人口以及个体随时间的变化之间存在很大差异。此外,我们对基因组内容和基因交换的分析表明,微病毒家族不携带可识别的辅助代谢基因,并且很少(如果有)在微病毒家族之间或与其细菌宿主之间进行水平基因转移。这些见解使微病毒分类学与其他噬菌体分类学的最新发展保持一致,并增加了对微病毒生物学的理解。微病毒是地球上最丰富的单链 DNA 噬菌体,也是人类肠道病毒组的重要组成部分。然而,由于当前分类排序的不足,对其生物学的生产性研究受到阻碍:微病毒被归入一个单一的科,并被视为一个整体,从而掩盖了它们多样性的程度,并导致比较研究很少。我们对微病毒多样性的研究定义了许多群体,其中大多数缺乏任何孤立的代表,这为未来的研究指明了高价值的目标。为了加快微病毒的发现和比较,我们开发了一个管道,能够快速简便地将新的微病毒基因组分类到定义明确的分类组中。这些改进为微病毒的生物学提供了新的见解,并强调了这些微型噬菌体与其大型双链 DNA 噬菌体竞争者之间的根本区别。