Department of Psychiatry, Virginia Commonwealth University, Richmond, VA, USA.
Department of Psychiatry, Texas A&M University, College Station, TX, USA.
Epigenetics. 2022 Dec;17(12):1753-1773. doi: 10.1080/15592294.2022.2079293. Epub 2022 Jun 13.
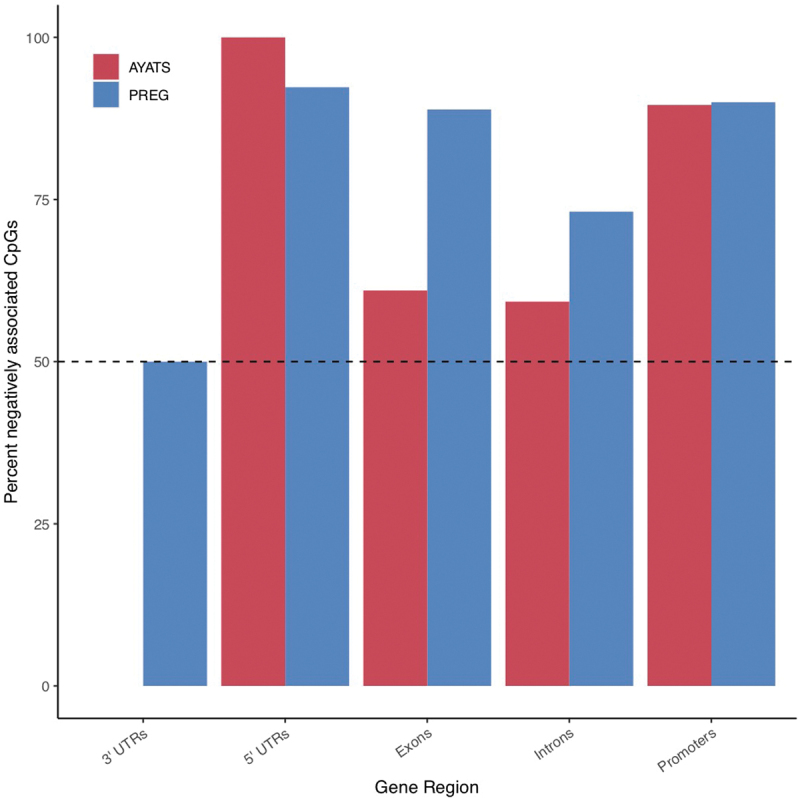
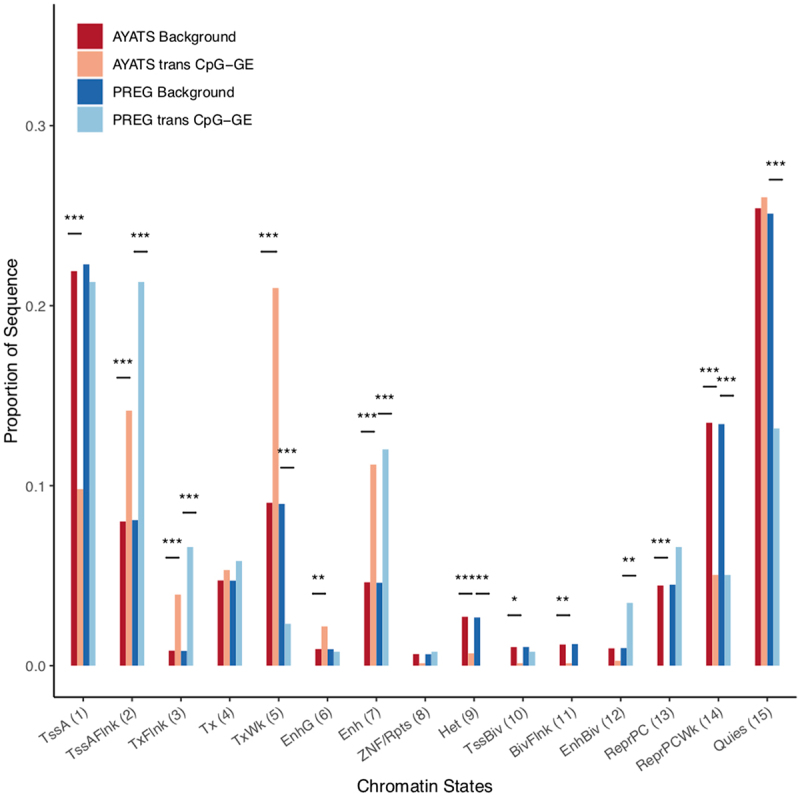
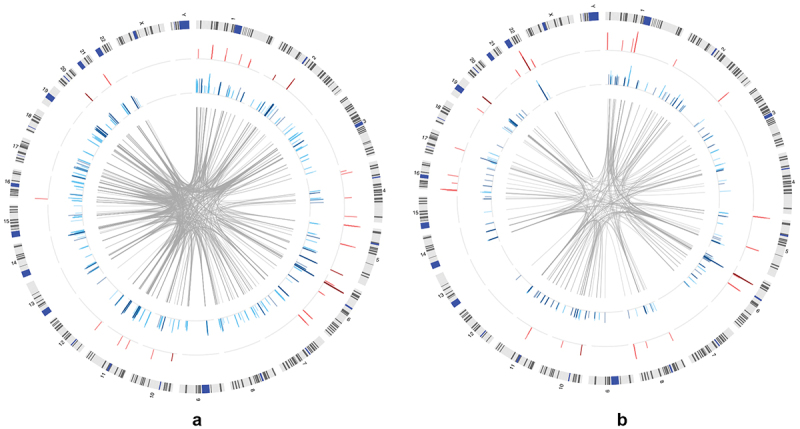
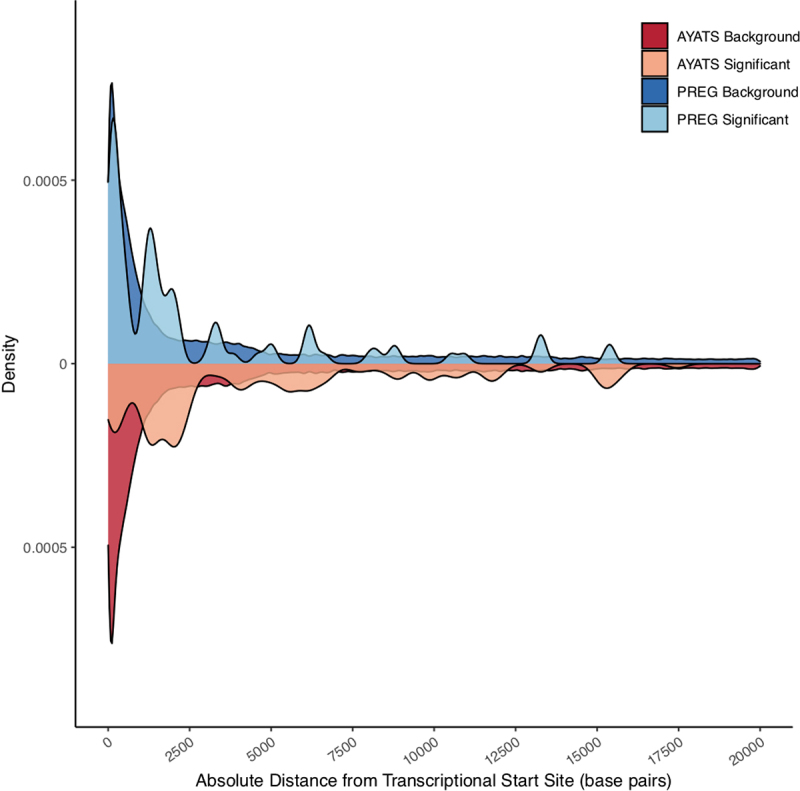
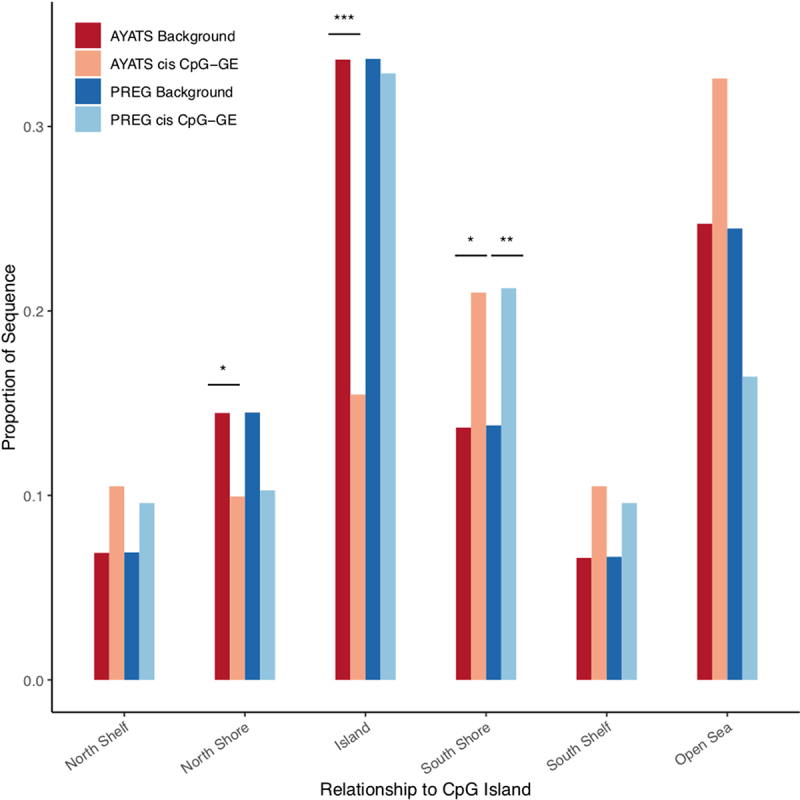
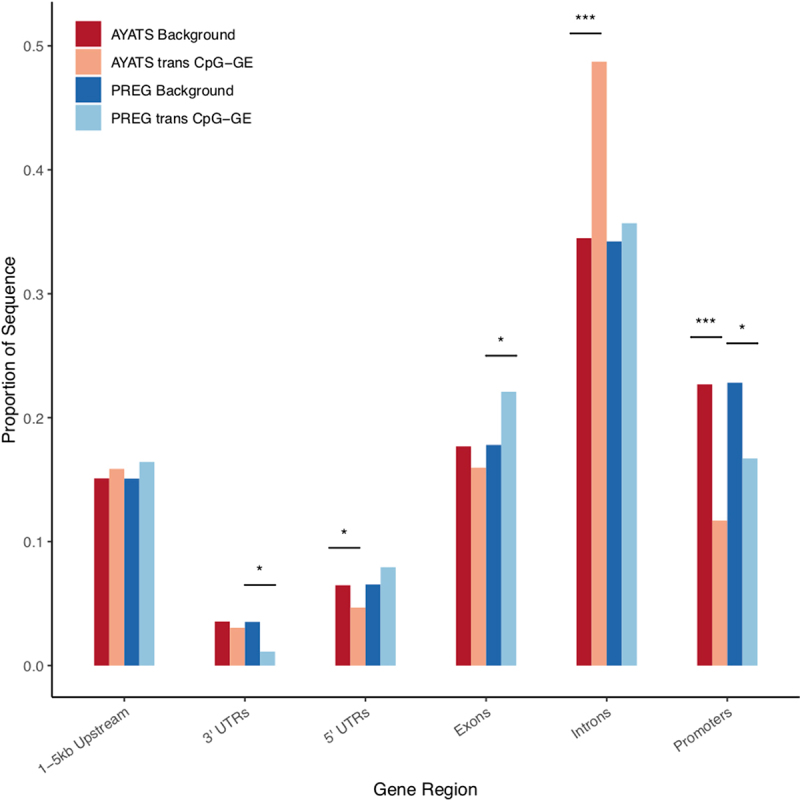
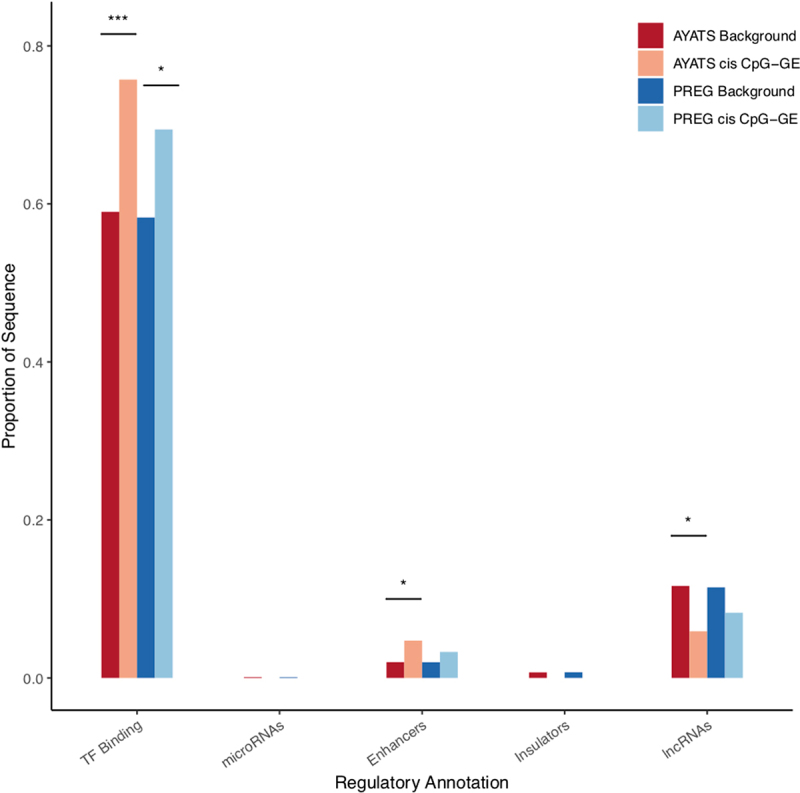
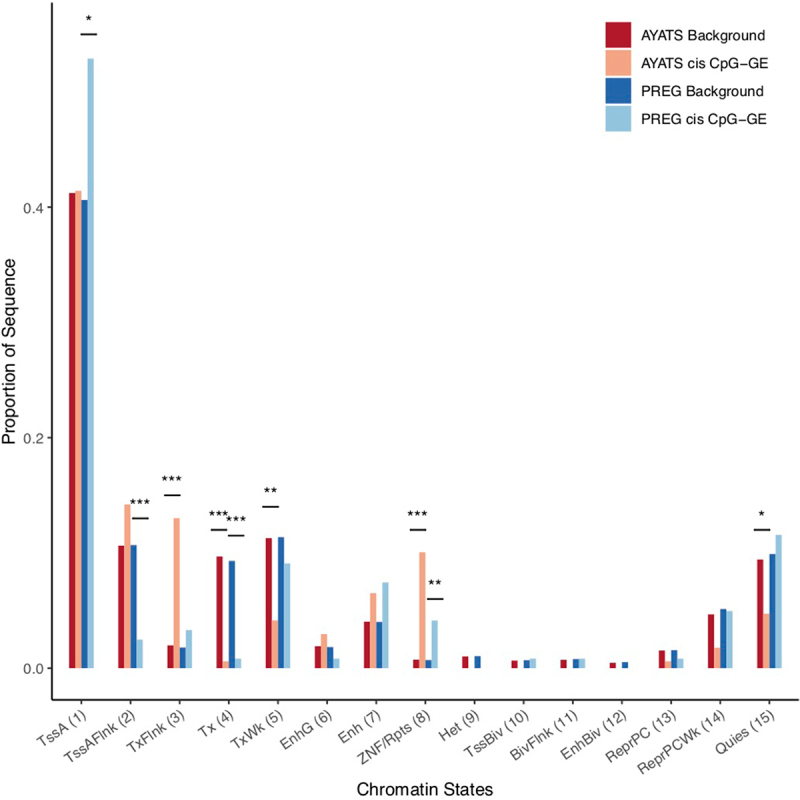
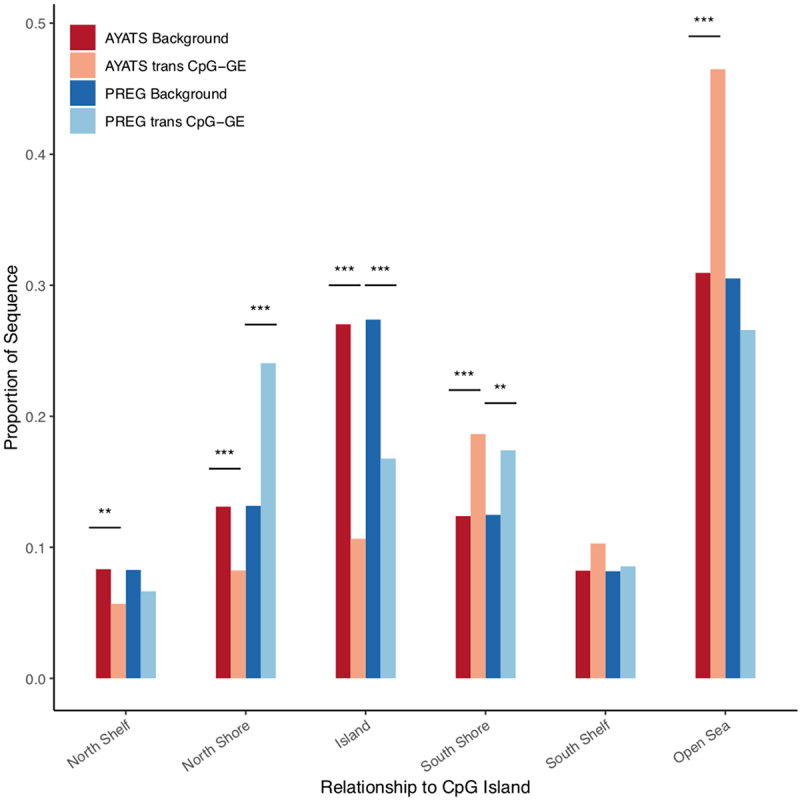
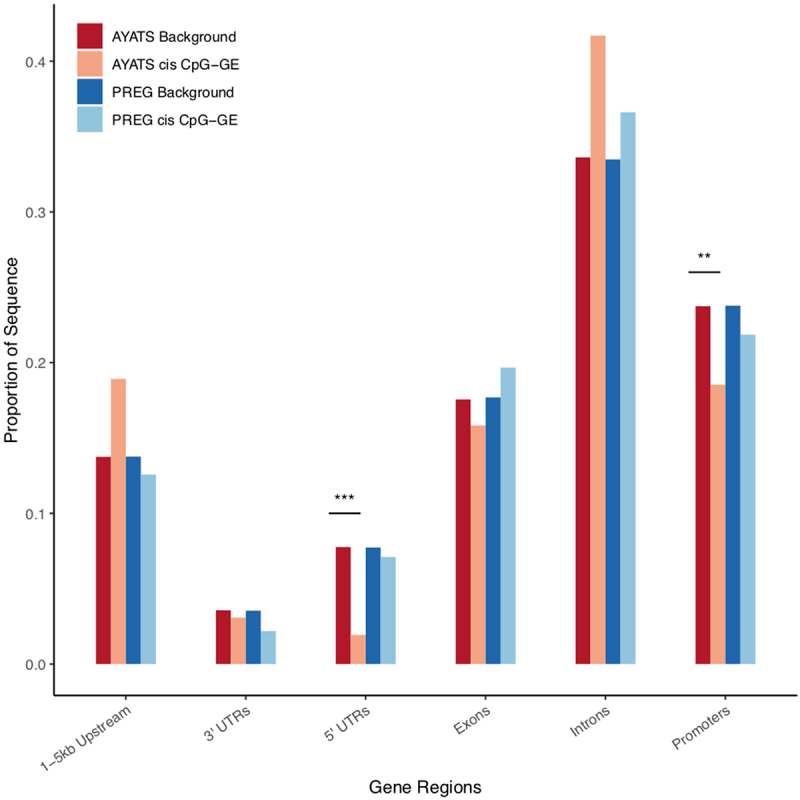
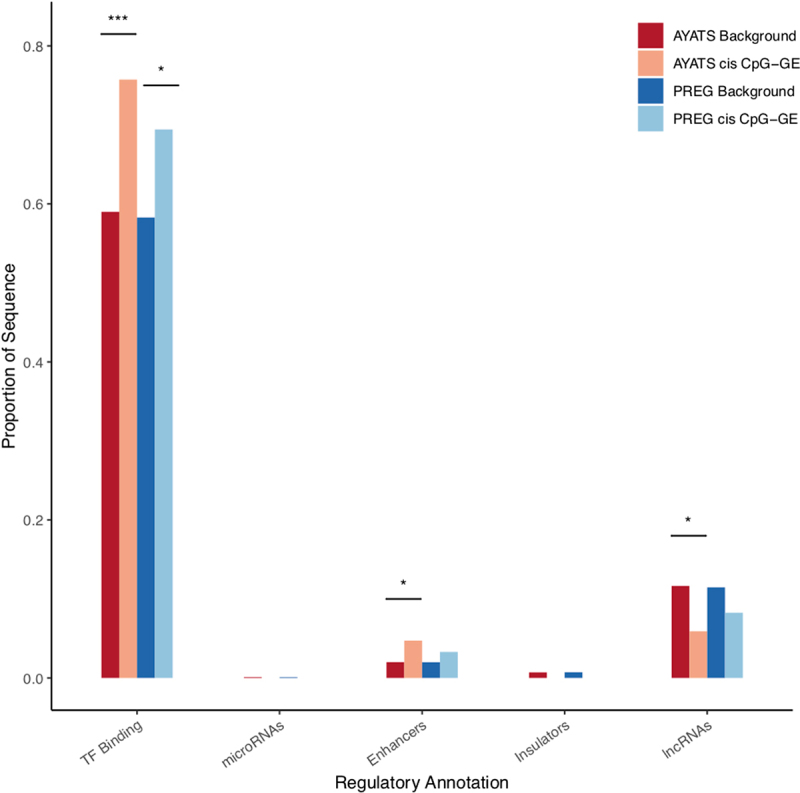
Although epigenome-wide association studies (EWAS) have been successful in identifying DNA methylation (DNAm) patterns associated with disease states, any further characterization of etiologic mechanisms underlying disease remains elusive. This knowledge gap does not originate from a lack of DNAm-trait associations, but rather stems from study design issues that affect the interpretability of EWAS results. Despite known limitations in predicting the function of a particular CpG site, most EWAS maintain the broad assumption that altered DNAm results in a concomitant change of transcription at the most proximal gene. This study integrated DNAm and gene expression (GE) measurements in two cohorts, the Adolescent and Young Adult Twin Study (AYATS) and the Pregnancy, Race, Environment, Genes (PREG) study, to improve the understanding of epigenomic regulatory mechanisms. CpG sites associated with GE in were enriched in areas of transcription factor binding and areas of intermediate-to-low CpG density. CpG sites associated with GE were also enriched in areas of known regulatory significance, including enhancer regions. These results highlight issues with restricting DNAm-transcript annotations to small genomic intervals and question the validity of assuming a DNAm-GE pathway. Based on these findings, the interpretation of EWAS results is limited in studies without multi-omic support and further research should identify genomic regions in which GE-associated DNAm is overrepresented. An in-depth characterization of GE-associated CpG sites could improve predictions of the downstream functional impact of altered DNAm and inform best practices for interpreting DNAm-trait associations generated by EWAS.
尽管全基因组关联研究(EWAS)已成功识别出与疾病状态相关的 DNA 甲基化(DNAm)模式,但对于疾病背后的病因机制仍缺乏进一步的认识。这一知识空白并非源于 DNAm 与特征之间关联的缺乏,而是源于影响 EWAS 结果可解释性的研究设计问题。尽管人们已知预测特定 CpG 位点功能存在局限性,但大多数 EWAS 仍然广泛假设 DNAm 的改变会导致最接近的基因转录随之发生变化。本研究在两个队列(青少年和年轻成人双胞胎研究(AYATS)和妊娠、种族、环境、基因(PREG)研究)中整合了 DNAm 和基因表达(GE)测量,以增进对表观基因组调控机制的理解。与 中的 GE 相关的 CpG 位点在转录因子结合区域和中低 CpG 密度区域富集。与 GE 相关的 CpG 位点也在已知的调控意义区域富集,包括增强子区域。这些结果突出了将 DNAm-转录注释限制在小基因组间隔的问题,并质疑了假设 DNAm-GE 途径的有效性。基于这些发现,在没有多组学支持的研究中,EWAS 结果的解释受到限制,进一步的研究应确定与 GE 相关的 DNAm 过度表达的基因组区域。对与 GE 相关的 CpG 位点进行深入表征可以改善对改变的 DNAm 下游功能影响的预测,并为解释 EWAS 生成的 DNAm-特征关联提供最佳实践。