Key Laboratory of Plant Molecular Physiology, Institute of Botany, Chinese Academy of Sciences, Beijing, 100093, China.
University of Chinese Academy of Sciences, Beijing, 100049, China.
BMC Plant Biol. 2022 Jun 13;22(1):288. doi: 10.1186/s12870-022-03677-8.
Wheat (Triticum aestivum L.) is an important cereal crop. Increasing grain yield for wheat is always a priority. Due to the complex genome of hexaploid wheat with 21 chromosomes, it is difficult to identify underlying genes by traditional genetic approach. The combination of genetics and omics analysis has displayed the powerful capability to identify candidate genes for major quantitative trait loci (QTLs), but such studies have rarely been carried out in wheat. In this study, candidate genes related to yield were predicted by a combined use of linkage mapping and weighted gene co-expression network analysis (WGCNA) in a recombinant inbred line population.
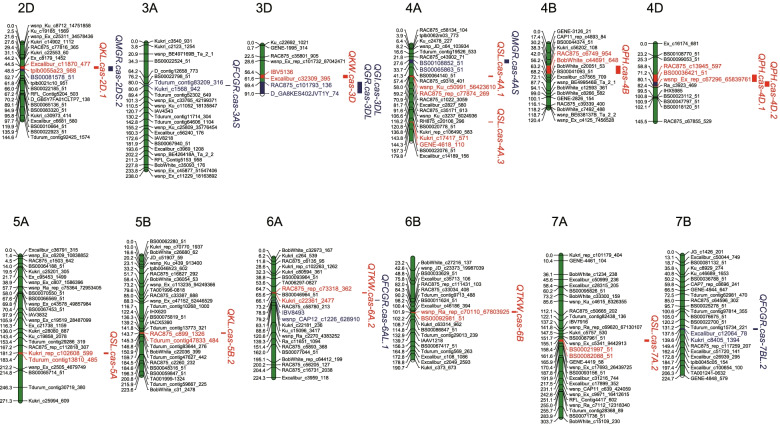
QTL mapping was performed for plant height (PH), spike length (SL) and seed traits. A total of 68 QTLs were identified for them, among which, 12 QTLs were stably identified across different environments. Using RNA sequencing, we scanned the 99,168 genes expression patterns of the whole spike for the recombinant inbred line population. By the combined use of QTL mapping and WGCNA, 29, 47, 20, 26, 54, 46 and 22 candidate genes were predicted for PH, SL, kernel length (KL), kernel width, thousand kernel weight, seed dormancy, and seed vigor, respectively. Candidate genes for different traits had distinct preferences. The known PH regulation genes Rht-B and Rht-D, and the known seed dormancy regulation genes TaMFT can be selected as candidate gene. Moreover, further experiment revealed that there was a SL regulatory QTL located in an interval of about 7 Mbp on chromosome 7A, named TaSL1, which also involved in the regulation of KL.
A combination of QTL mapping and WGCNA was applied to predicted wheat candidate genes for PH, SL and seed traits. This strategy will facilitate the identification of candidate genes for related QTLs in wheat. In addition, the QTL TaSL1 that had multi-effect regulation of KL and SL was identified, which can be used for wheat improvement. These results provided valuable molecular marker and gene information for fine mapping and cloning of the yield-related trait loci in the future.
小麦(Triticum aestivum L.)是一种重要的谷类作物。提高小麦的产量一直是首要任务。由于六倍体小麦的基因组复杂,有 21 条染色体,传统的遗传方法很难鉴定潜在的基因。遗传学和组学分析的结合已经显示出鉴定主要数量性状位点(QTL)候选基因的强大能力,但在小麦中这样的研究很少进行。在这项研究中,通过连锁作图和加权基因共表达网络分析(WGCNA)的结合,在重组自交系群体中预测了与产量相关的候选基因。
对株高(PH)、穗长(SL)和种子性状进行了 QTL 作图。共鉴定到 68 个 QTL,其中 12 个 QTL在不同环境下稳定鉴定。利用 RNA 测序,我们扫描了重组自交系群体中整个穗的 99168 个基因的表达模式。通过 QTL 作图和 WGCNA 的结合,分别预测了 PH、SL、千粒重、种子休眠、种子活力 29、47、20、26、46 和 22 个候选基因。不同性状的候选基因有明显的偏好。已知的 PH 调节基因 Rht-B 和 Rht-D 以及已知的种子休眠调节基因 TaMFT 可作为候选基因。此外,进一步的实验表明,在 7A 染色体上大约 7 Mbp 的区间存在一个 SL 调节 QTL,命名为 TaSL1,它也参与了 KL 的调节。
将 QTL 作图和 WGCNA 相结合,预测了 PH、SL 和种子性状的小麦候选基因。该策略将有助于鉴定小麦相关 QTL 的候选基因。此外,还鉴定到了一个具有 KL 和 SL 多效调节作用的 QTL TaSL1,可用于小麦改良。这些结果为未来产量相关性状位点的精细定位和克隆提供了有价值的分子标记和基因信息。