Wan Mingyu, Yue Han, Notarangelo Jaime, Liu Hongfu, Che Fanglin
Department of Chemical Engineering, University of Massachusetts Lowell, Lowell 01854, United States.
Michtom School of Computer Science, Brandeis University, Waltham, Massachusetts 02453, United States.
JACS Au. 2022 Jun 2;2(6):1338-1349. doi: 10.1021/jacsau.2c00003. eCollection 2022 Jun 27.
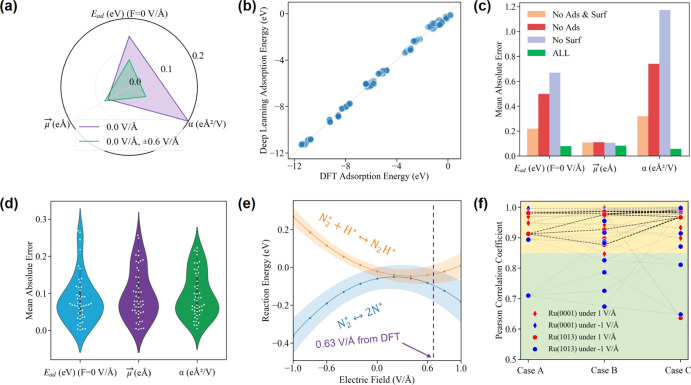
External electric fields can modify binding energies of reactive surface species and enhance catalytic performance of heterogeneously catalyzed reactions. In this work, we used density functional theory (DFT) calculations-assisted and accelerated by a deep learning algorithm-to investigate the extent to which ruthenium-catalyzed ammonia synthesis would benefit from application of such external electric fields. This strategy allows us to determine which electronic properties control a molecule's degree of interaction with external electric fields. Our results show that (1) field-dependent adsorption/reaction energies are closely correlated to the dipole moments of intermediates over the surface, (2) a positive field promotes ammonia synthesis by lowering the overall energetics and decreasing the activation barriers of the potential rate-limiting steps (e.g., NH hydrogenation) over Ru, (3) a positive field (>0.6 V/Å) favors the reaction mechanism by avoiding kinetically unfavorable N≡N bond dissociation over Ru(1013), and (4) local adsorption environments (i.e., dipole moments of the intermediates in the gas phase, surface defects, and surface coverage of intermediates) influence the resulting surface adsorbates' dipole moments and further modify field-dependent reaction energetics. The deep learning algorithm developed here accelerates field-dependent energy predictions with acceptable accuracies by five orders of magnitudes compared to DFT alone and has the capacity of transferability, which can predict field-dependent energetics of other catalytic surfaces with high-quality performance using little training data.
外部电场可以改变反应性表面物种的结合能,并提高多相催化反应的催化性能。在这项工作中,我们使用了由深度学习算法辅助和加速的密度泛函理论(DFT)计算,以研究钌催化氨合成在应用这种外部电场时将受益的程度。这种策略使我们能够确定哪些电子性质控制分子与外部电场的相互作用程度。我们的结果表明:(1)与场相关的吸附/反应能量与表面中间体的偶极矩密切相关;(2)正电场通过降低整体能量和降低钌上潜在限速步骤(例如NH加氢)的活化能垒来促进氨合成;(3)正电场(>0.6 V/Å)通过避免在Ru(1013)上动力学不利的N≡N键解离而有利于反应机理;(4)局部吸附环境(即气相中中间体的偶极矩、表面缺陷和中间体的表面覆盖率)影响所得表面吸附物的偶极矩,并进一步改变与场相关的反应能量。与单独的DFT相比,这里开发的深度学习算法将具有可接受精度的与场相关的能量预测加速了五个数量级,并且具有可转移性,能够使用少量训练数据以高质量性能预测其他催化表面的与场相关的能量。