Center for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN, 37831, USA.
Department of Chemistry and Biochemistry, Queens College of the City University of New York, Queens, NY, 11367, USA.
Nat Commun. 2021 Jan 4;12(1):88. doi: 10.1038/s41467-020-20342-6.
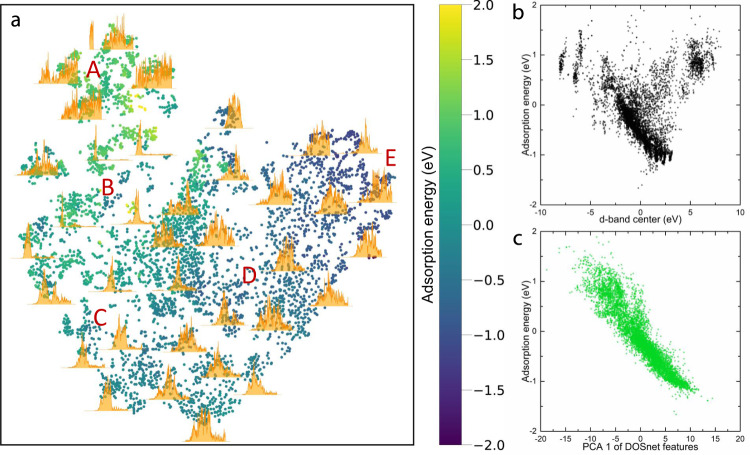
Materials databases generated by high-throughput computational screening, typically using density functional theory (DFT), have become valuable resources for discovering new heterogeneous catalysts, though the computational cost associated with generating them presents a crucial roadblock. Hence there is a significant demand for developing descriptors or features, in lieu of DFT, to accurately predict catalytic properties, such as adsorption energies. Here, we demonstrate an approach to predict energies using a convolutional neural network-based machine learning model to automatically obtain key features from the electronic density of states (DOS). The model, DOSnet, is evaluated for a diverse set of adsorbates and surfaces, yielding a mean absolute error on the order of 0.1 eV. In addition, DOSnet can provide physically meaningful predictions and insights by predicting responses to external perturbations to the electronic structure without additional DFT calculations, paving the way for the accelerated discovery of materials and catalysts by exploration of the electronic space.
利用高通量计算筛选生成的材料数据库,通常使用密度泛函理论 (DFT),已成为发现新型多相催化剂的有价值资源,尽管生成这些数据库所涉及的计算成本是一个关键障碍。因此,人们强烈需要开发描述符或特征,以替代 DFT,从而准确预测催化性能,如吸附能。在这里,我们展示了一种使用基于卷积神经网络的机器学习模型来预测能量的方法,该模型可以自动从电子态密度 (DOS) 中获取关键特征。该模型 DOSnet 针对各种吸附物和表面进行了评估,其在吸附能上的平均绝对误差约为 0.1 eV。此外,DOSnet 可以通过预测对电子结构的外部扰动的响应来提供有物理意义的预测和见解,而无需进行额外的 DFT 计算,为通过探索电子空间来加速发现材料和催化剂铺平了道路。