Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125.
Entos, Inc., Los Angeles, CA 90027.
Proc Natl Acad Sci U S A. 2022 Aug 2;119(31):e2205221119. doi: 10.1073/pnas.2205221119. Epub 2022 Jul 28.
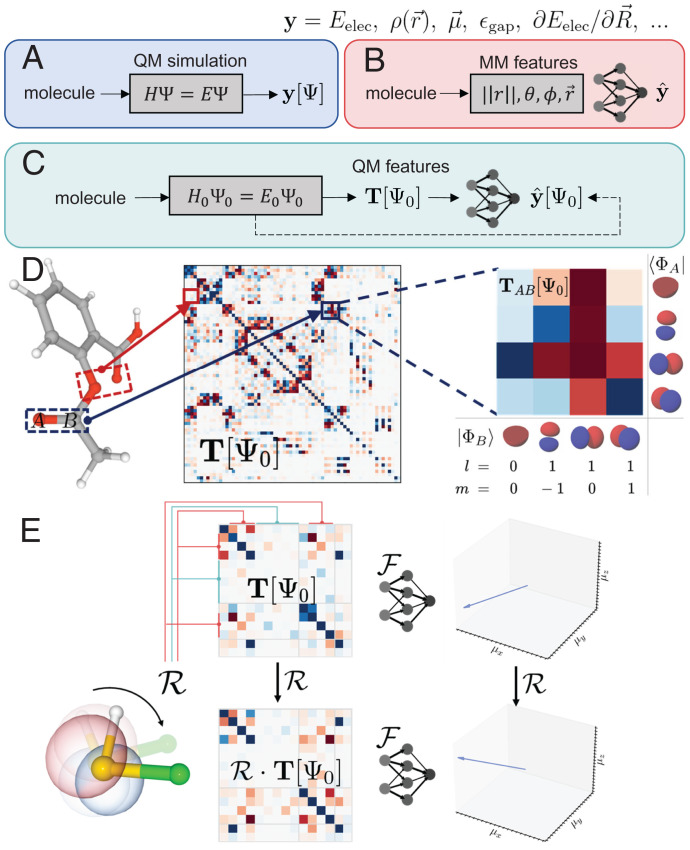
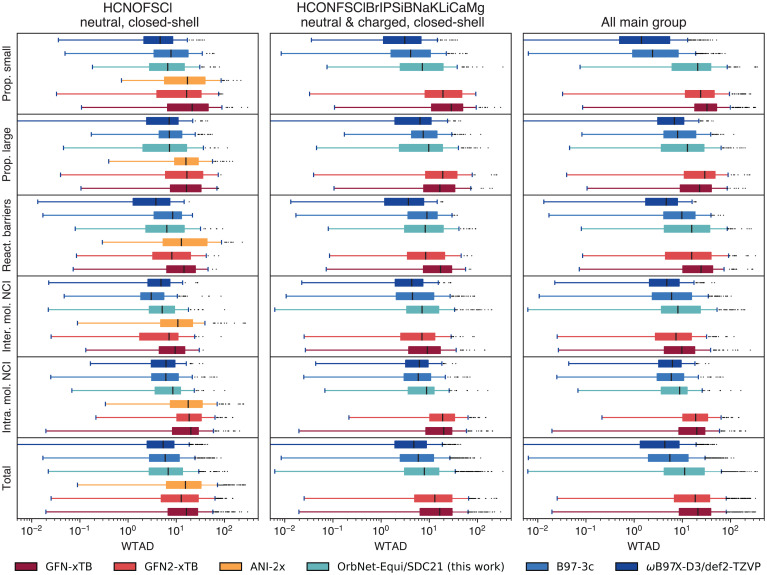
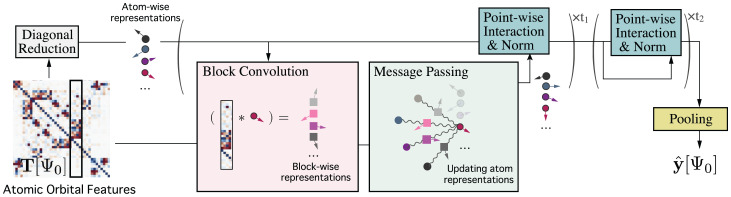
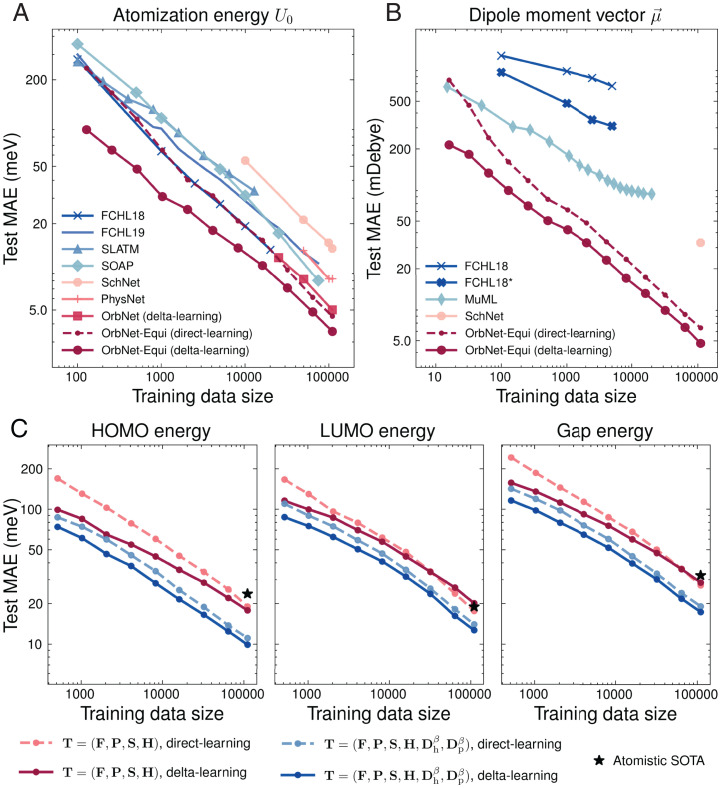
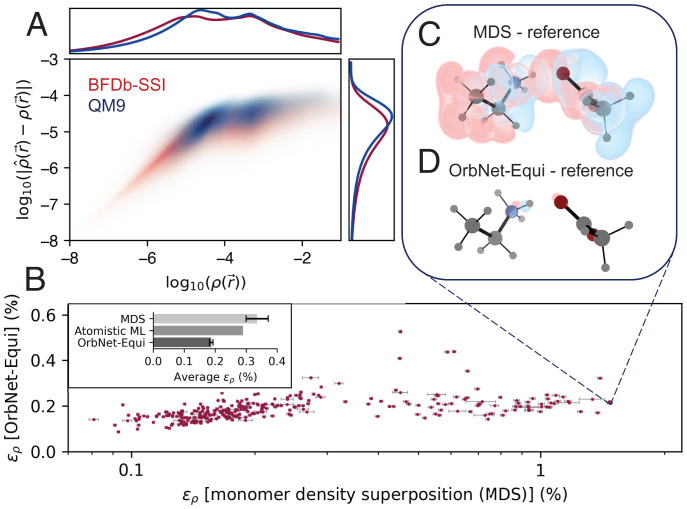
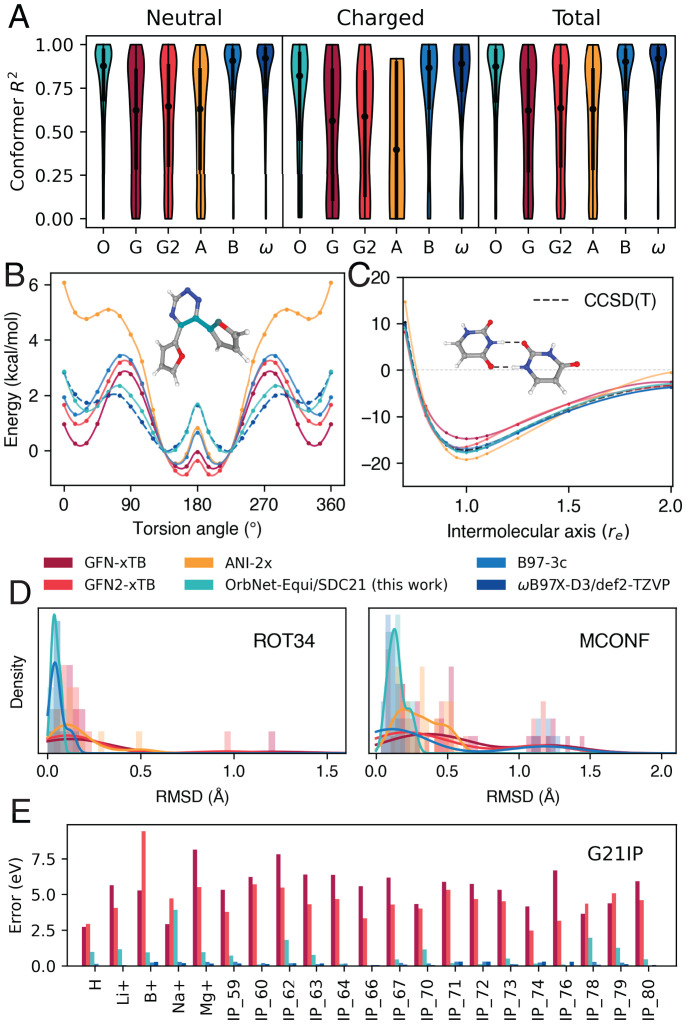
Predicting electronic energies, densities, and related chemical properties can facilitate the discovery of novel catalysts, medicines, and battery materials. However, existing machine learning techniques are challenged by the scarcity of training data when exploring unknown chemical spaces. We overcome this barrier by systematically incorporating knowledge of molecular electronic structure into deep learning. By developing a physics-inspired equivariant neural network, we introduce a method to learn molecular representations based on the electronic interactions among atomic orbitals. Our method, OrbNet-Equi, leverages efficient tight-binding simulations and learned mappings to recover high-fidelity physical quantities. OrbNet-Equi accurately models a wide spectrum of target properties while being several orders of magnitude faster than density functional theory. Despite only using training samples collected from readily available small-molecule libraries, OrbNet-Equi outperforms traditional semiempirical and machine learning-based methods on comprehensive downstream benchmarks that encompass diverse main-group chemical processes. Our method also describes interactions in challenging charge-transfer complexes and open-shell systems. We anticipate that the strategy presented here will help to expand opportunities for studies in chemistry and materials science, where the acquisition of experimental or reference training data is costly.
预测电子能量、密度和相关化学性质可以促进新型催化剂、药物和电池材料的发现。然而,当探索未知化学空间时,现有的机器学习技术受到训练数据稀缺的挑战。我们通过系统地将分子电子结构知识纳入深度学习来克服这一障碍。通过开发一种受物理启发的等变神经网络,我们引入了一种基于原子轨道间电子相互作用学习分子表示的方法。我们的方法 OrbNet-Equi 利用高效的紧束缚模拟和学习映射来恢复高保真的物理量。OrbNet-Equi 可以准确地模拟广泛的目标性质,同时速度比密度泛函理论快几个数量级。尽管仅使用从小分子库中收集的训练样本,但 OrbNet-Equi 在涵盖多种主族化学过程的全面下游基准测试中优于传统的半经验和基于机器学习的方法。我们的方法还描述了具有挑战性的电荷转移配合物和开壳系统中的相互作用。我们预计,这里提出的策略将有助于扩大化学和材料科学领域的研究机会,在这些领域中,获取实验或参考训练数据的成本很高。