Grupo de Diseño de Productos y Procesos (GDPP), Department of Chemical and Food Engineering, Universidad de los Andes, Bogotá, Colombia.
Naturalius SAS, Bogotá, Colombia.
Sci Rep. 2022 Aug 18;12(1):14030. doi: 10.1038/s41598-022-17204-0.
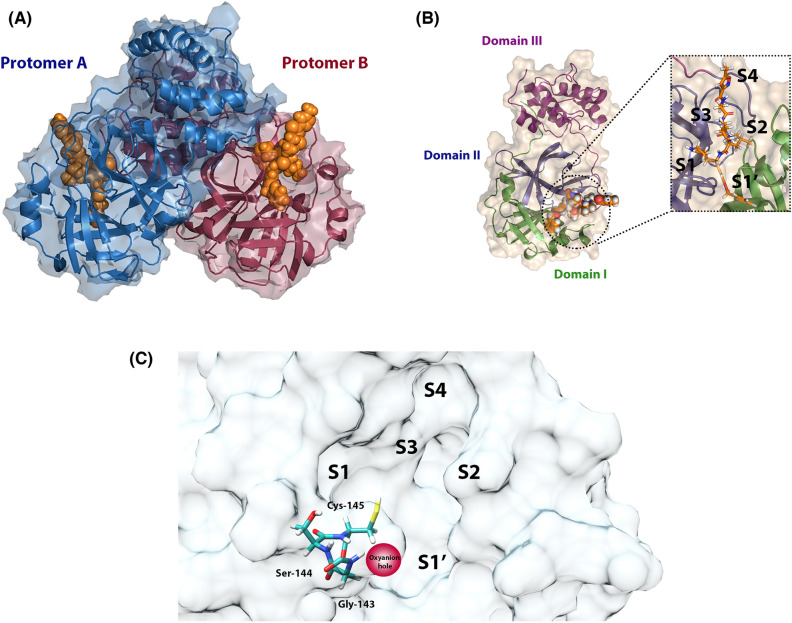
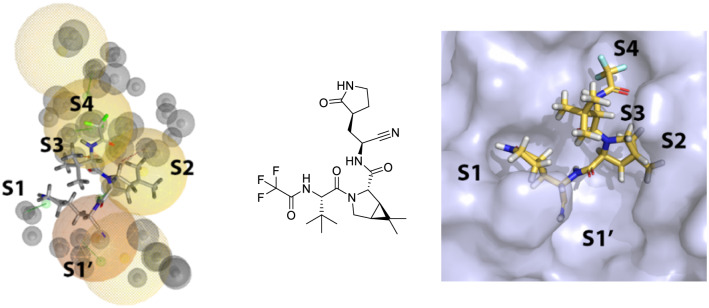
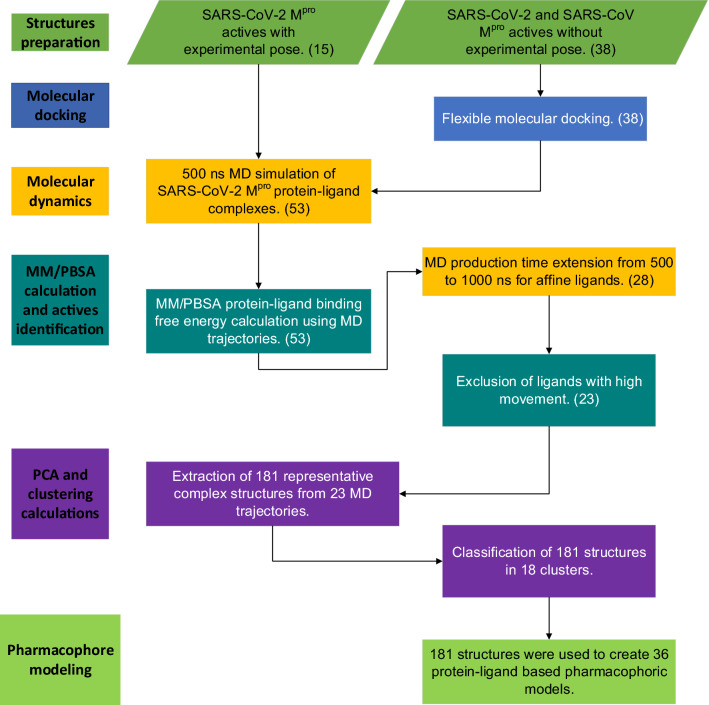
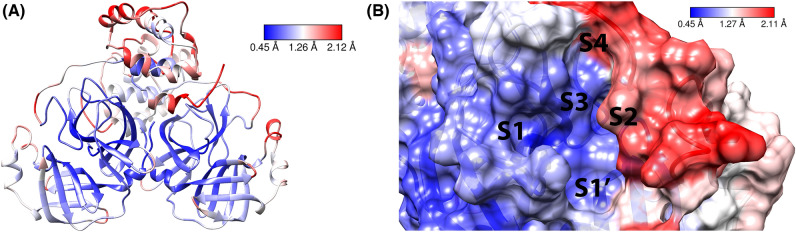
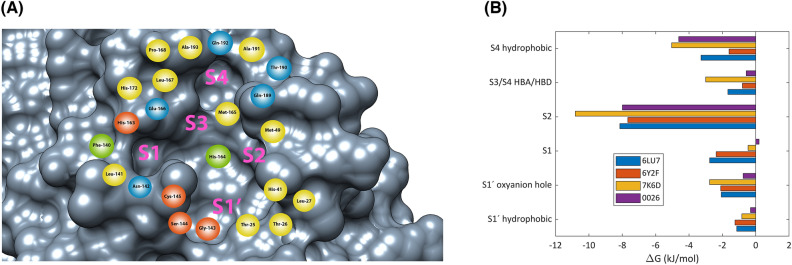
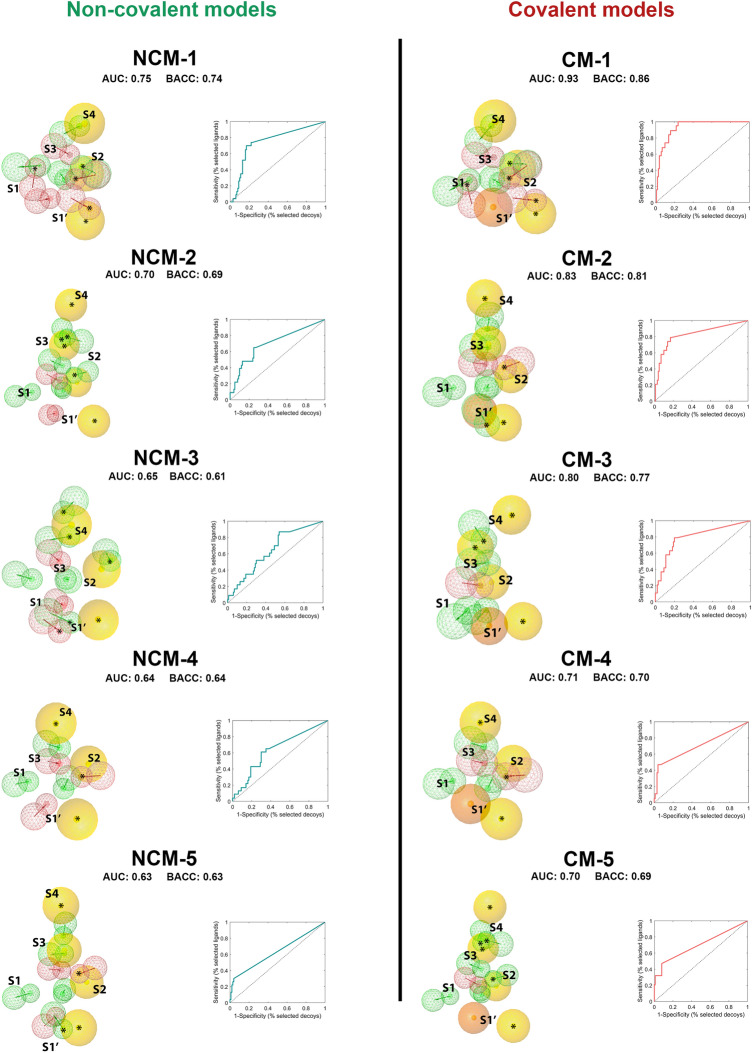
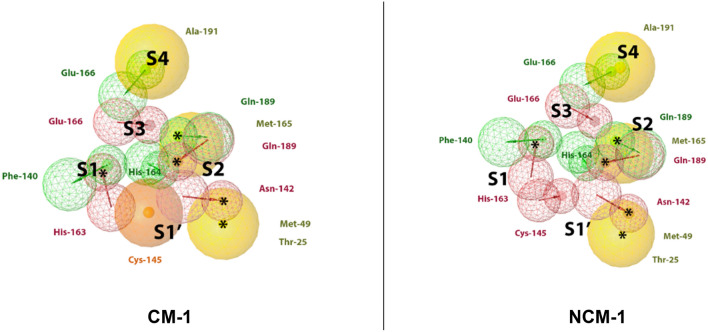
As the world enters its second year of the pandemic caused by SARS-CoV-2, intense efforts have been directed to develop an effective diagnosis, prevention, and treatment strategies. One promising drug target to design COVID-19 treatments is the SARS-CoV-2 M. To date, a comparative understanding of M dynamic stereoelectronic interactions with either covalent or non-covalent inhibitors (depending on their interaction with a pocket called S1' or oxyanion hole) has not been still achieved. In this study, we seek to fill this knowledge gap using a cascade in silico protocol of docking, molecular dynamics simulations, and MM/PBSA in order to elucidate pharmacophore models for both types of inhibitors. After docking and MD analysis, a set of complex-based pharmacophore models was elucidated for covalent and non-covalent categories making use of the residue bonding point feature. The highest ranked models exhibited ROC-AUC values of 0.93 and 0.73, respectively for each category. Interestingly, we observed that the active site region of M protein-ligand complex undergoes large conformational changes, especially within the S2 and S4 subsites. The results reported in this article may be helpful in virtual screening (VS) campaigns to guide the design and discovery of novel small-molecule therapeutic agents against SARS-CoV-2 M protein.
随着由 SARS-CoV-2 引起的大流行进入第二年,人们已经投入了大量的努力来开发有效的诊断、预防和治疗策略。设计 COVID-19 治疗方法的一个有前途的药物靶点是 SARS-CoV-2 M。迄今为止,人们尚未对 M 与共价或非共价抑制剂(取决于它们与称为 S1'或氧阴离子穴的口袋的相互作用)的动态立体电子相互作用进行比较性理解。在这项研究中,我们使用对接、分子动力学模拟和 MM/PBSA 的级联计算方案来填补这一知识空白,以阐明这两种抑制剂的药效团模型。在对接和 MD 分析之后,利用残基键合点特征,为共价和非共价两类阐明了一组基于复合物的药效团模型。每个类别中得分最高的模型的 ROC-AUC 值分别为 0.93 和 0.73。有趣的是,我们观察到 M 蛋白-配体复合物的活性位点区域经历了较大的构象变化,特别是在 S2 和 S4 亚位点内。本文报道的结果可能有助于虚拟筛选(VS)活动,以指导针对 SARS-CoV-2 M 蛋白的新型小分子治疗剂的设计和发现。