Department of Statistics, Florida State University, Tallahassee, FL, 32306-4330, USA.
BMC Bioinformatics. 2022 Sep 8;23(1):368. doi: 10.1186/s12859-022-04912-7.
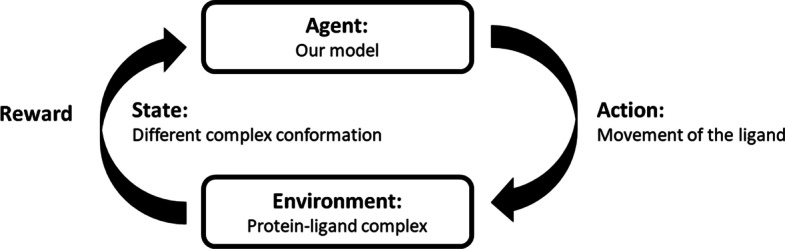
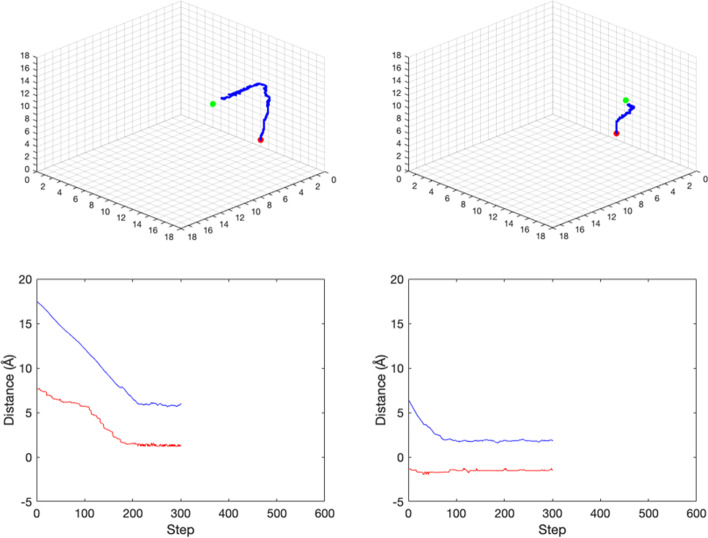
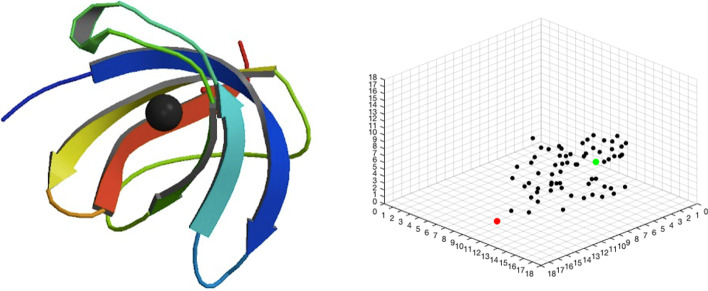
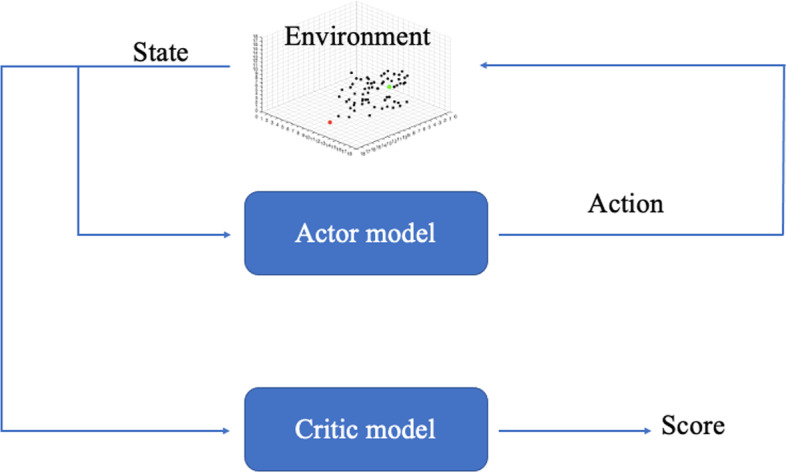
Protein ligand docking is an indispensable tool for computational prediction of protein functions and screening drug candidates. Despite significant progress over the past two decades, it is still a challenging problem, characterized by the still limited understanding of the energetics between proteins and ligands, and the vast conformational space that has to be searched to find a satisfactory solution. In this project, we developed a novel reinforcement learning (RL) approach, the asynchronous advantage actor-critic model (A3C), to address the protein ligand docking problem. The overall framework consists of two models. During the search process, the agent takes an action selected by the actor model based on the current location. The critic model then evaluates this action and predict the distance between the current location and true binding site. Experimental results showed that in both single- and multi-atom cases, our model improves binding site prediction substantially compared to a naïve model. For the single-atom ligand, copper ion (Cu), the model predicted binding sites have a median root-mean-square-deviation (RMSD) of 2.39 Å to the true binding sites when starting from random starting locations. For the multi-atom ligand, sulfate ion (SO), the predicted binding sites have a median RMSD of 3.82 Å to the true binding sites. The ligand-specific models built in this study can be used in solvent mapping studies and the RL framework can be readily scaled up to larger and more diverse sets of ligands.
蛋白质配体对接是计算预测蛋白质功能和筛选药物候选物不可或缺的工具。尽管在过去的二十年中取得了重大进展,但它仍然是一个具有挑战性的问题,其特点是对蛋白质和配体之间的能量仍然理解有限,并且必须搜索广阔的构象空间才能找到令人满意的解决方案。在这个项目中,我们开发了一种新的强化学习(RL)方法,异步优势演员-评论家模型(A3C),来解决蛋白质配体对接问题。整体框架由两个模型组成。在搜索过程中,代理根据当前位置从演员模型中选择一个动作。然后,评论家模型评估此动作并预测当前位置与真实结合位点之间的距离。实验结果表明,在单原子和多原子情况下,与简单模型相比,我们的模型大大提高了结合位点的预测。对于单原子配体,铜离子(Cu),当从随机起始位置开始时,模型预测的结合位点的中值均方根偏差(RMSD)为 2.39 Å。对于多原子配体,硫酸根离子(SO),预测的结合位点的中值 RMSD 为 3.82 Å。本研究中构建的配体特异性模型可用于溶剂映射研究,RL 框架可轻松扩展到更大和更多样化的配体集。