Kandel Jeevan, Tayara Hilal, Chong Kil To
Graduate School of Integrated Energy-AI, Jeonbuk National University, Jeonju, 54896, South Korea.
School of International Engineering and Science, Jeonbuk National University, Jeonju, 54896, South Korea.
J Cheminform. 2021 Sep 8;13(1):65. doi: 10.1186/s13321-021-00547-7.
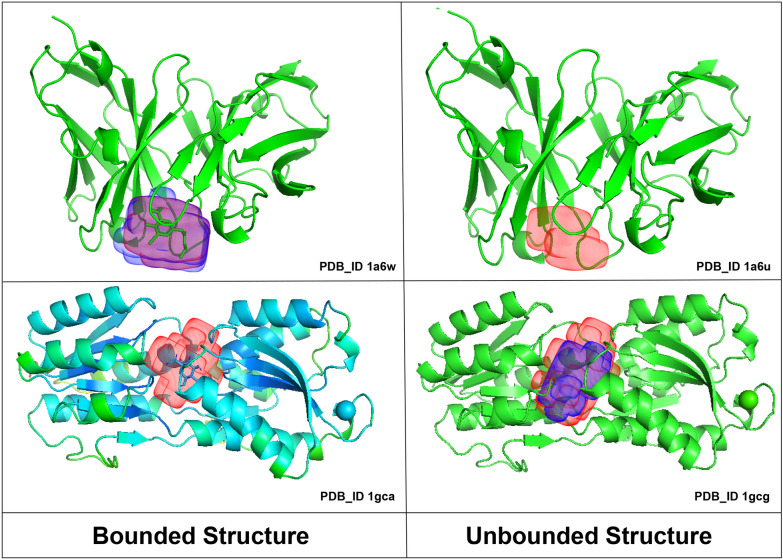
Predicting protein-ligand binding sites is a fundamental step in understanding the functional characteristics of proteins, which plays a vital role in elucidating different biological functions and is a crucial step in drug discovery. A protein exhibits its true nature after binding to its interacting molecule known as a ligand that binds only in the favorable binding site of the protein structure. Different computational methods exploiting the features of proteins have been developed to identify the binding sites in the protein structure, but none seems to provide promising results, and therefore, further investigation is required.
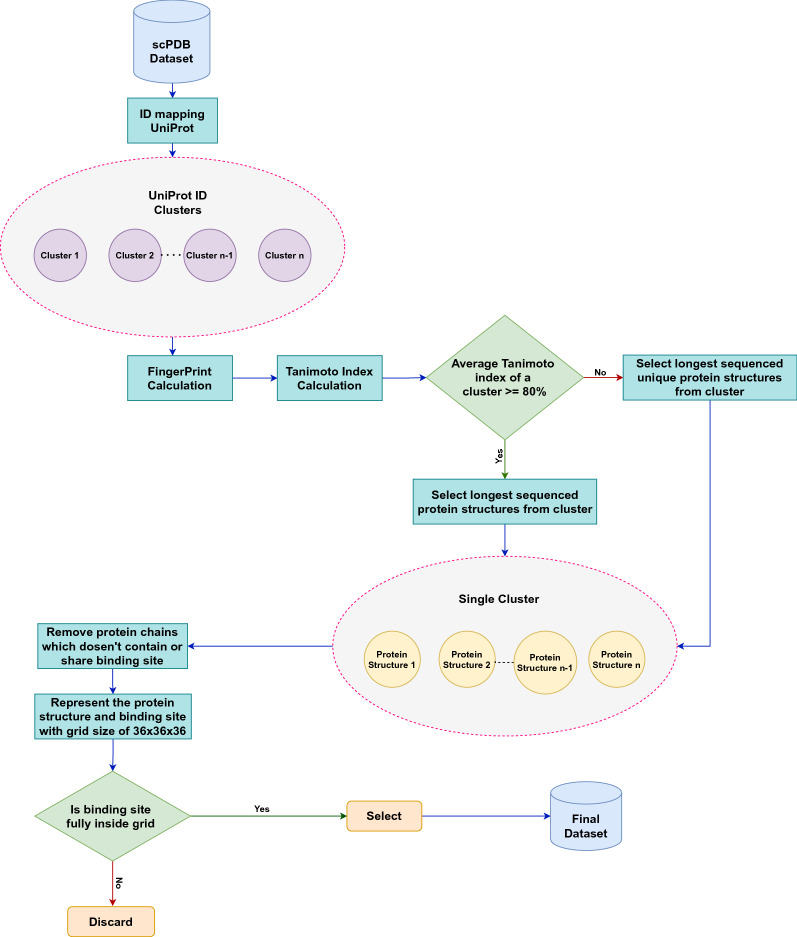
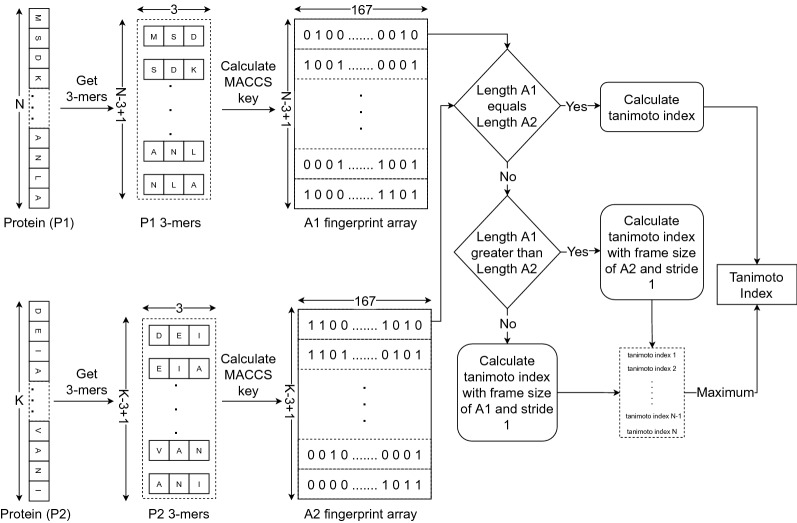
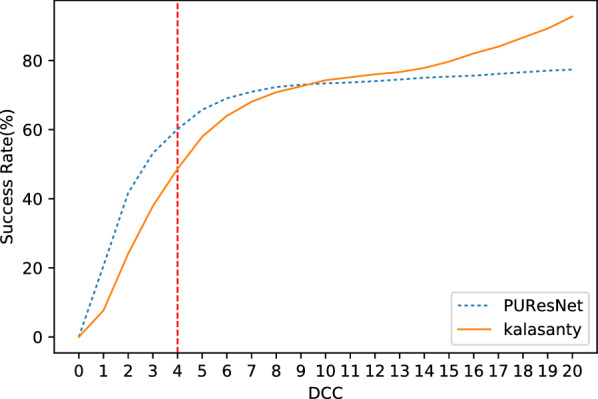
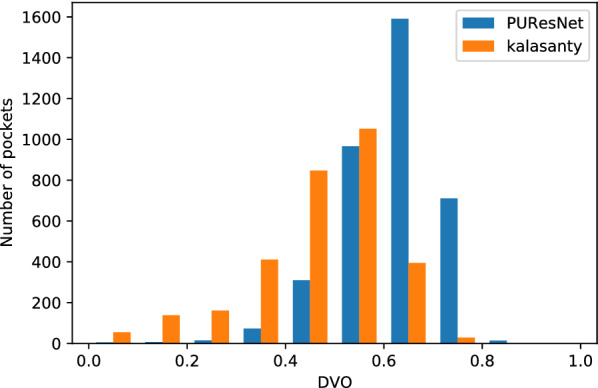
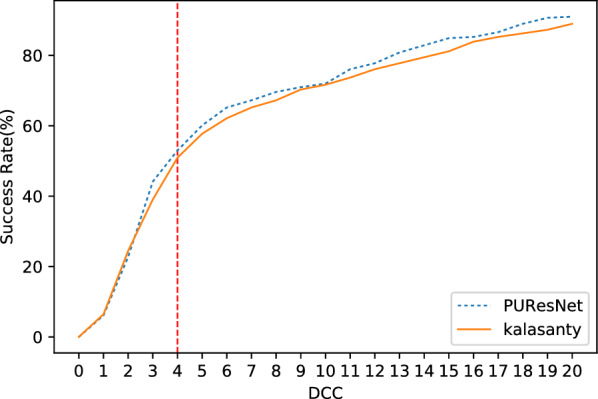
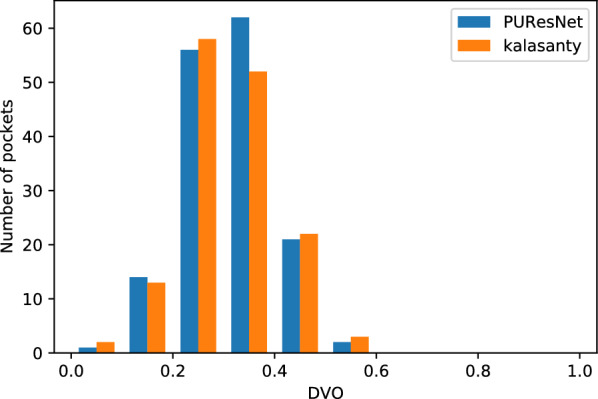
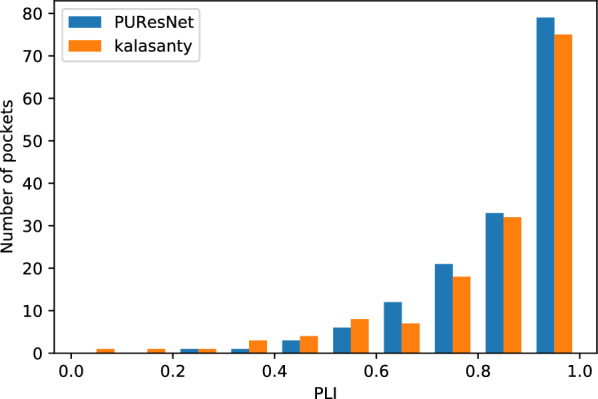
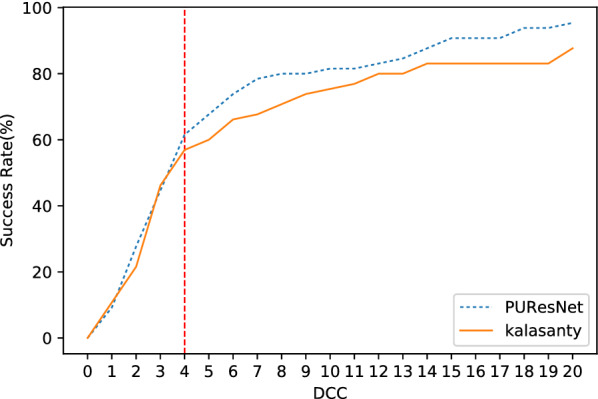
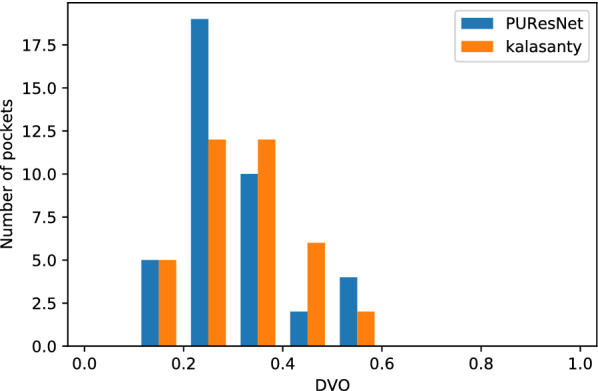
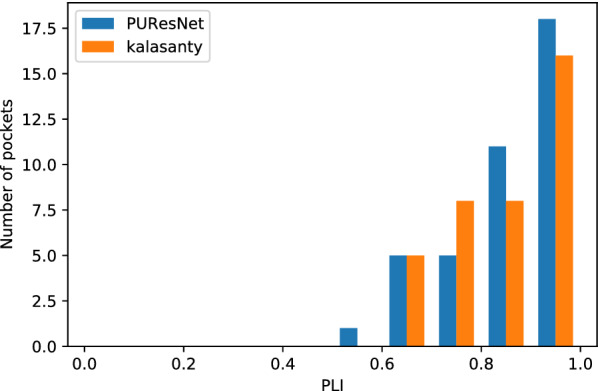
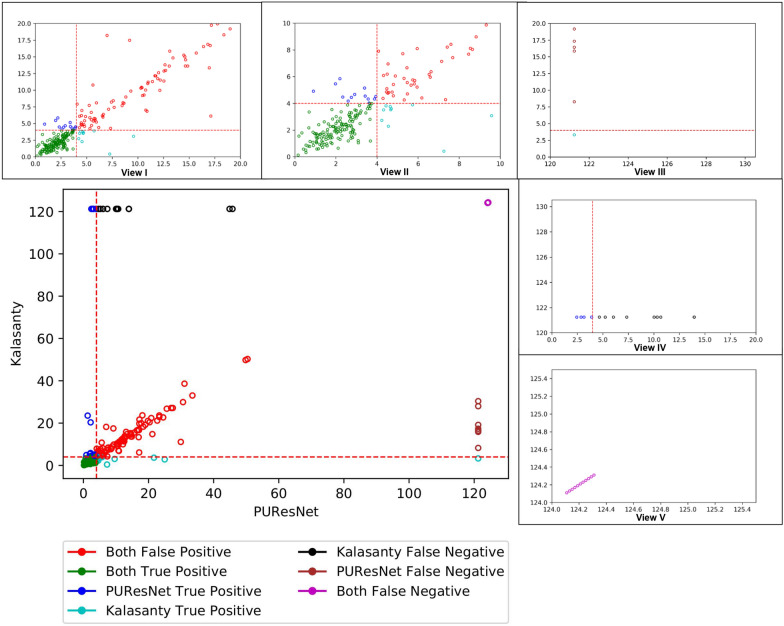
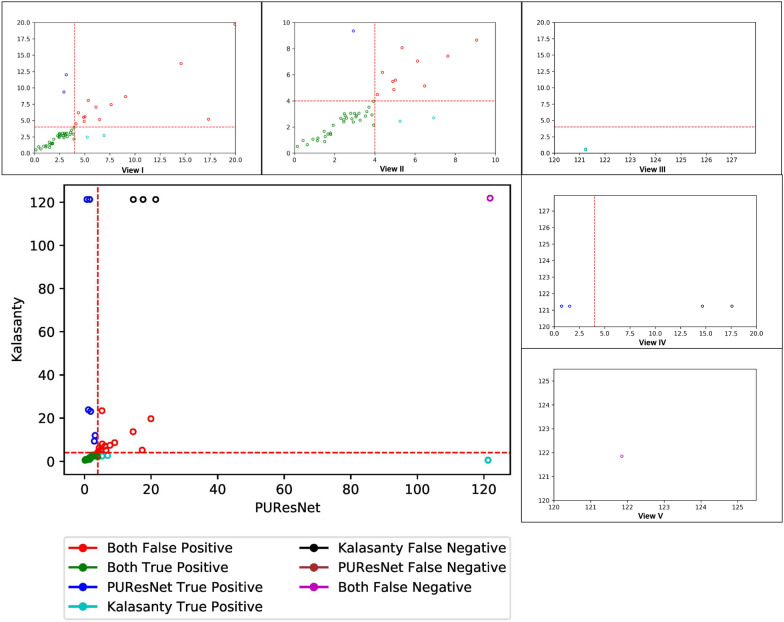
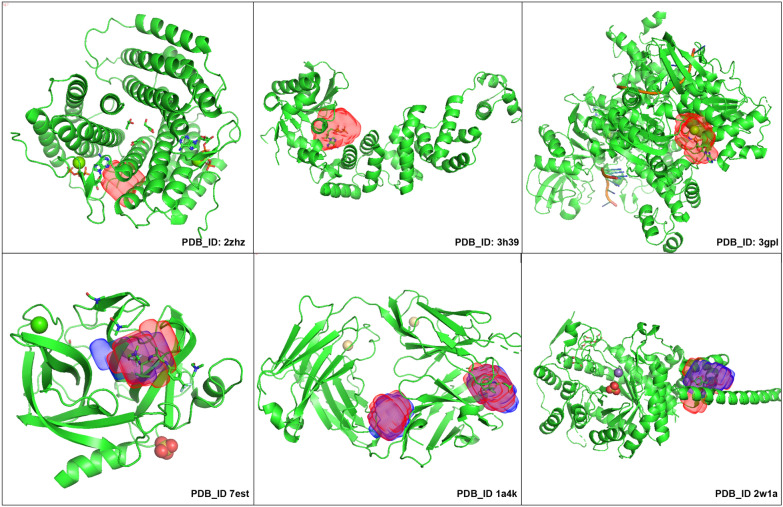
In this study, we present a deep learning model PUResNet and a novel data cleaning process based on structural similarity for predicting protein-ligand binding sites. From the whole scPDB (an annotated database of druggable binding sites extracted from the Protein DataBank) database, 5020 protein structures were selected to address this problem, which were used to train PUResNet. With this, we achieved better and justifiable performance than the existing methods while evaluating two independent sets using distance, volume and proportion metrics.
预测蛋白质 - 配体结合位点是理解蛋白质功能特性的基本步骤,这在阐明不同生物学功能中起着至关重要的作用,并且是药物发现中的关键一步。蛋白质与称为配体的相互作用分子结合后展现其真实性质,而配体仅在蛋白质结构的有利结合位点结合。已经开发了利用蛋白质特征的不同计算方法来识别蛋白质结构中的结合位点,但似乎没有一个能提供有前景的结果,因此需要进一步研究。
在本研究中,我们提出了一种深度学习模型PUResNet和一种基于结构相似性的新型数据清理过程,用于预测蛋白质 - 配体结合位点。从整个scPDB(从蛋白质数据库中提取的可成药结合位点注释数据库)数据库中,选择了5020个蛋白质结构来解决这个问题,这些结构用于训练PUResNet。通过这种方式,在使用距离、体积和比例指标评估两个独立数据集时,我们取得了比现有方法更好且合理的性能。