Bai Jianhao, Wan Zhongqi, Zhang Yuanyuan, Wang Tianyu, Xue Yawen, Peng Qing
Department of Ophthalmology, Shanghai Tenth People's Hospital of Tongji University, Tongji University School of Medicine, Shanghai, China.
Front Microbiol. 2022 Aug 23;13:926926. doi: 10.3389/fmicb.2022.926926. eCollection 2022.
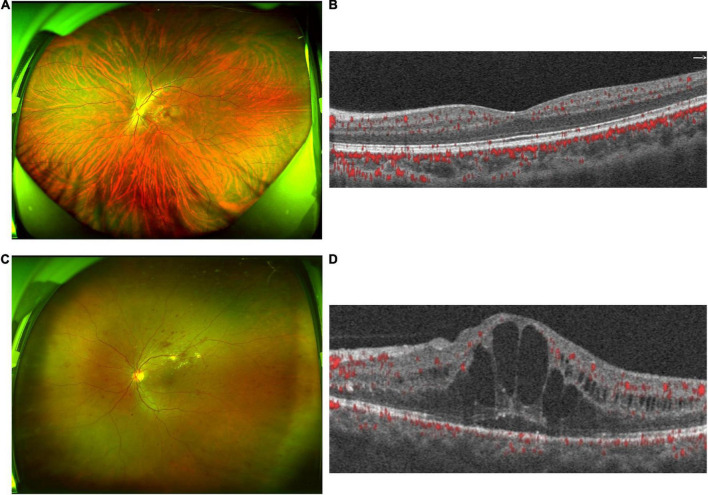
Diabetic retinopathy (DR) is one of the most common complications of type 2 diabetes mellitus. The current study investigates the composition, structure, and function of gut microbiota in DR patients and explores the correlation between gut microbiota and clinical characteristics of DR.
A total of 50 stool samples were collected from 50 participants, including 25 DR patients and 25 healthy controls (HCs). 16S ribosomal RNA gene sequencing was used to analyze the gut microbial composition in these two groups. DNA was extracted from the fecal samples using the MiSeq platform.
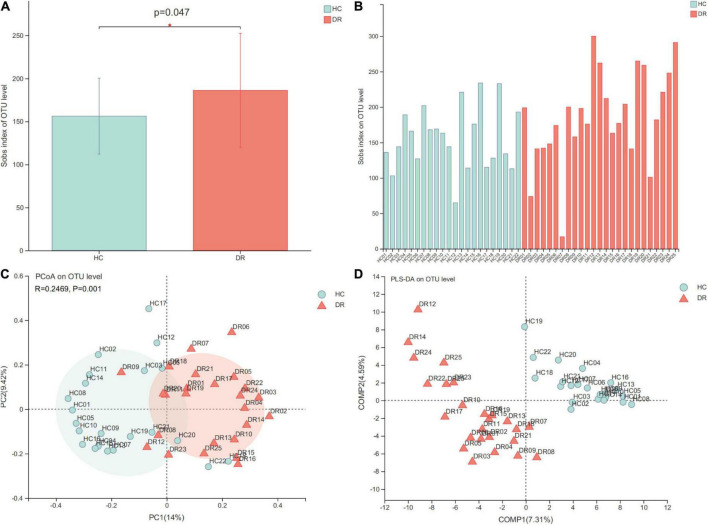
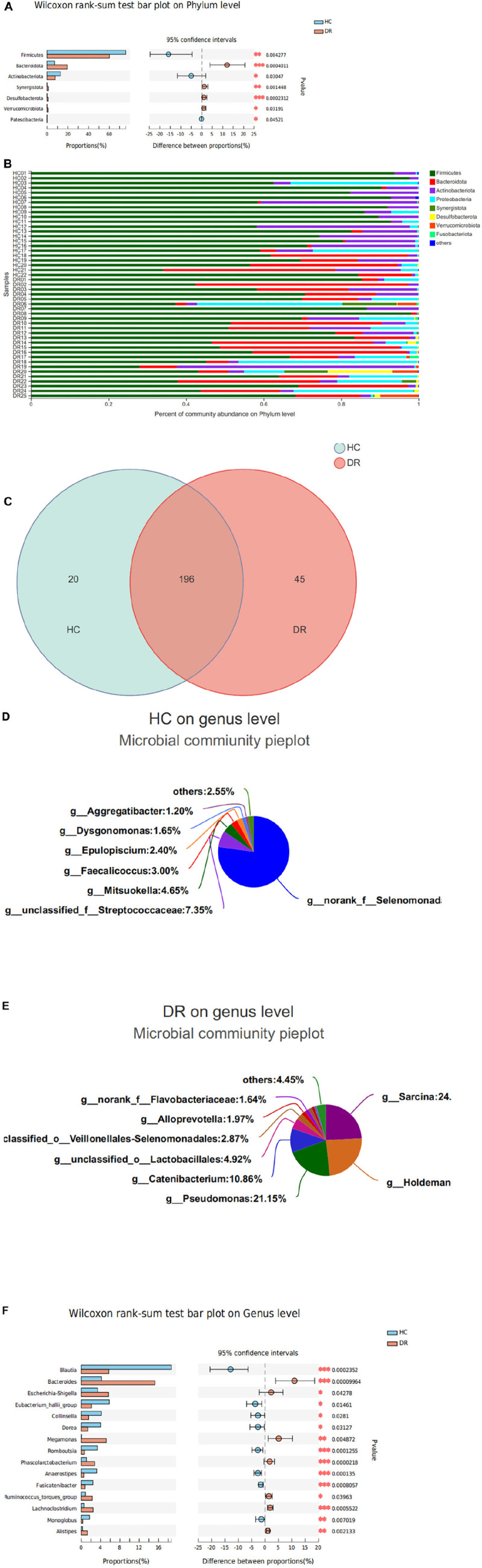
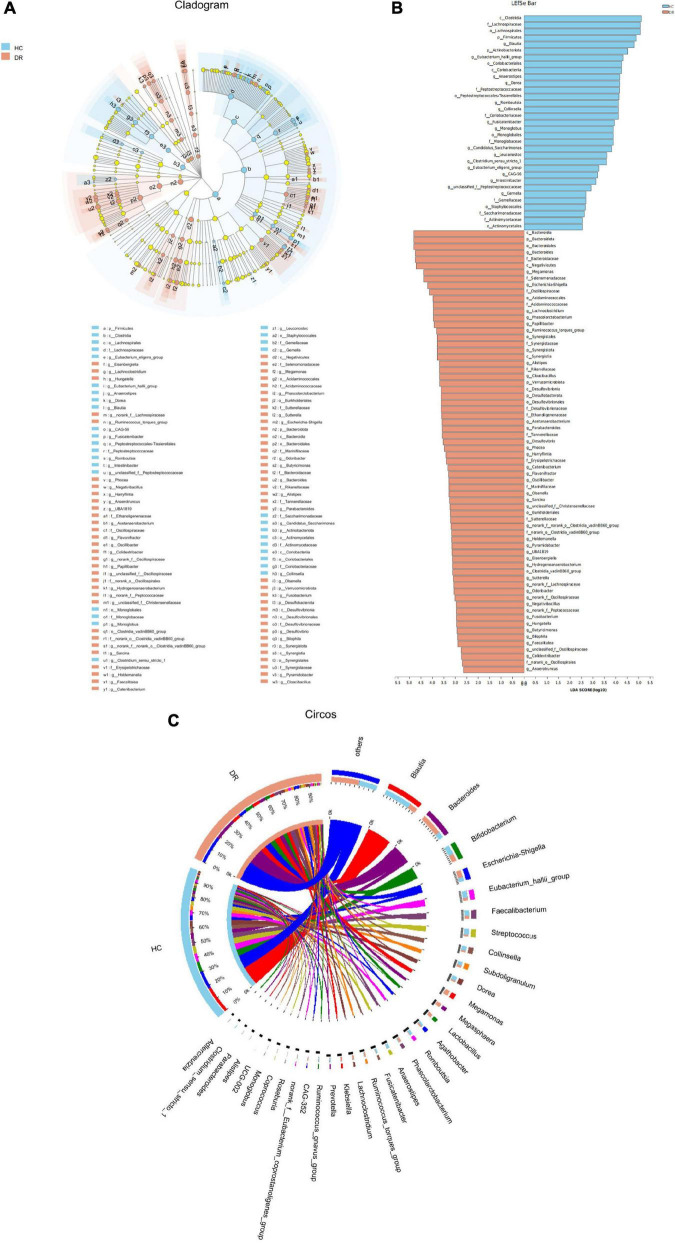
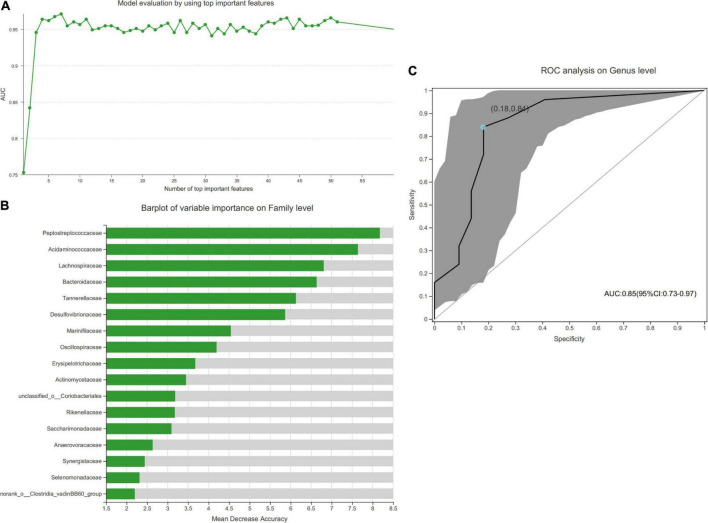
The microbial structure and composition of DR patients were different from that of HCs. The microbial richness of gut microbiota in DR was higher than that of normal individuals. The alterations of microbiome of DR patients were associated with disrupted Firmicutes, Bacteroidetes, Synergistota, and Desulfobacterota phyla. In addition, increased levels of , , _group, , and , and decreased levels of , _group, , , , , and genera were observed in the DR groups. Additionally, a stochastic forest model was developed to identify a set of biomarkers with seven bacterial genera that can differentiate patients with DR from those HC. The microbial communities exhibited varied functions in these two groups because of the alterations of the above-mentioned bacterial genera.
The altered composition and function of gut microbiota in DR patients indicated that gut microbiome could be used as non-invasive biomarkers, improve clinical diagnostic methods, and identify putative therapeutic targets for DR.
糖尿病视网膜病变(DR)是2型糖尿病最常见的并发症之一。本研究调查了DR患者肠道微生物群的组成、结构和功能,并探讨了肠道微生物群与DR临床特征之间的相关性。
从50名参与者中收集了50份粪便样本,其中包括25名DR患者和25名健康对照(HC)。采用16S核糖体RNA基因测序分析两组的肠道微生物组成。使用MiSeq平台从粪便样本中提取DNA。
DR患者的微生物结构和组成与HC不同。DR患者肠道微生物群的丰富度高于正常个体。DR患者微生物组的改变与厚壁菌门、拟杆菌门、协同菌门和脱硫杆菌门的破坏有关。此外,在DR组中观察到[具体菌属1]、[具体菌属2]、[具体菌属3]_组、[具体菌属4]、[具体菌属5]水平升高,以及[具体菌属6]、[具体菌属7]_组、[具体菌属8]、[具体菌属9]、[具体菌属10]、[具体菌属11]水平降低。此外,开发了一种随机森林模型,以识别一组具有七个细菌属的生物标志物,这些生物标志物可以区分DR患者和HC。由于上述细菌属的改变,两组中的微生物群落表现出不同的功能。
DR患者肠道微生物群的组成和功能改变表明,肠道微生物组可作为非侵入性生物标志物,改善临床诊断方法,并确定DR的潜在治疗靶点。