Valerieva Anna, Longhurst Hilary J
Department of Allergology, Medical University of Sofia, Sofia, Bulgaria.
Department of Immunology, Auckland District Health Board, and Department of Medicine, University of Auckland, Auckland, New Zealand.
Front Allergy. 2022 Sep 12;3:952233. doi: 10.3389/falgy.2022.952233. eCollection 2022.
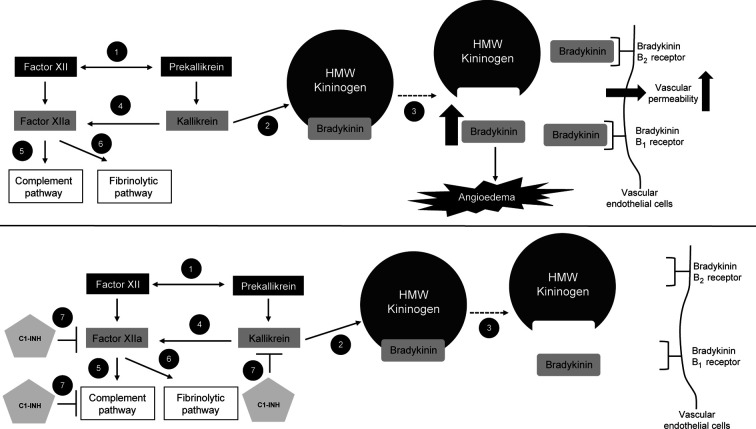
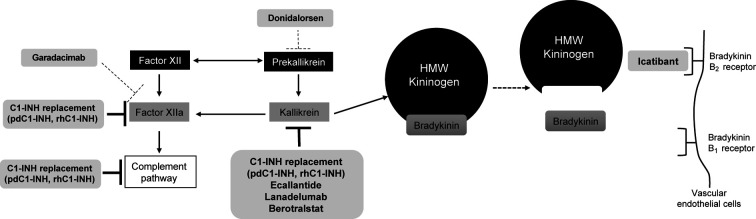
Hereditary angioedema (HAE) is a rare disease caused by mutations in the gene. This results in deficient or dysfunctional C1 esterase inhibitor (C1-INH) and affects multiple proteases involved in the complement, contact-system, coagulation, and fibrinolytic pathways. Current options for the treatment and prevention of HAE attacks include treating all affected pathways direct C1-INH replacement therapy; or specifically targeting components of the contact activation system, in particular by blocking the bradykinin B receptor (B2R) or inhibiting plasma kallikrein, to prevent bradykinin generation. Intravenously administered plasma-derived C1-INH (pdC1-INH) and recombinant human C1-INH have demonstrated efficacy and safety for treatment of HAE attacks, although time to onset of symptom relief varied among trials, specific agents, and dosing regimens. Data from retrospective and observational analyses support that short-term prophylaxis with intravenous C1-INH products can help prevent HAE attacks in patients undergoing medical or dental procedures. Long-term prophylaxis with intravenous or subcutaneous pdC1-INH significantly decreased the HAE attack rate vs. placebo, although breakthrough attacks were observed. Pathway-specific therapies for the management of HAE include the B2R antagonist icatibant and plasma kallikrein inhibitors ecallantide, lanadelumab, and berotralstat. Icatibant, administered for treatment of angioedema attacks, reduced B2R-mediated vascular permeability and, compared with placebo, reduced the time to initial symptom improvement. Plasma kallikrein inhibitors, such as ecallantide, block the binding site of kallikrein to prevent cleavage of high molecular weight kininogen and subsequent bradykinin generation. Ecallantide was shown to be efficacious for HAE attacks and is licensed for this indication in the United States, but the labeling recommends that only health care providers administer treatment because of the risk of anaphylaxis. In addition to C1-INH replacement therapy, the plasma kallikrein inhibitors lanadelumab and berotralstat are recommended as first-line options for long-term prophylaxis and have demonstrated marked reductions in HAE attack rates. Investigational therapies, including the activated factor XII inhibitor garadacimab and an antisense oligonucleotide targeting plasma prekallikrein messenger RNA (donidalorsen), have shown promise as long-term prophylaxis. Given the requirement of lifelong management for HAE, further research is needed to determine how best to individualize optimal treatments for each patient.
遗传性血管性水肿(HAE)是一种由基因突变引起的罕见疾病。这会导致C1酯酶抑制剂(C1-INH)缺乏或功能失调,并影响参与补体、接触系统、凝血和纤溶途径的多种蛋白酶。目前治疗和预防HAE发作的选择包括针对所有受影响途径进行治疗——直接进行C1-INH替代疗法;或特异性靶向接触激活系统的成分,特别是通过阻断缓激肽B2受体(B2R)或抑制血浆激肽释放酶,以防止缓激肽生成。静脉注射血浆源性C1-INH(pdC1-INH)和重组人C1-INH已证明对治疗HAE发作有效且安全,尽管不同试验、特定药物和给药方案的症状缓解起效时间有所不同。回顾性和观察性分析的数据支持,静脉注射C1-INH产品进行短期预防可帮助预防接受医疗或牙科手术患者的HAE发作。与安慰剂相比,静脉或皮下注射pdC1-INH进行长期预防可显著降低HAE发作率,尽管仍观察到突破性发作。用于管理HAE的途径特异性疗法包括B2R拮抗剂艾替班特和血浆激肽释放酶抑制剂依库珠单抗、拉那单抗和贝罗司他。用于治疗血管性水肿发作的艾替班特可降低B2R介导的血管通透性,与安慰剂相比,缩短了初始症状改善时间。血浆激肽释放酶抑制剂,如依库珠单抗,可阻断激肽释放酶的结合位点,防止高分子量激肽原的裂解及随后缓激肽的生成。依库珠单抗已证明对HAE发作有效,在美国已获该适应症许可,但标签建议仅由医疗保健提供者进行治疗,因为存在过敏反应风险。除C1-INH替代疗法外,血浆激肽释放酶抑制剂拉那单抗和贝罗司他被推荐作为长期预防的一线选择,并已证明可显著降低HAE发作率。研究性疗法,包括活化因子XII抑制剂加拉西单抗和靶向血浆前激肽释放酶信使核糖核酸的反义寡核苷酸(多尼达森),已显示出作为长期预防的潜力。鉴于HAE需要终身管理,需要进一步研究以确定如何为每位患者最佳地个体化优化治疗。