Theoretical Chemistry, Vrije Universiteit, De Boelelaan 1083, NL-1081 HVAmsterdam, The Netherlands.
J Chem Theory Comput. 2022 Nov 8;18(11):6779-6793. doi: 10.1021/acs.jctc.2c00531. Epub 2022 Oct 6.
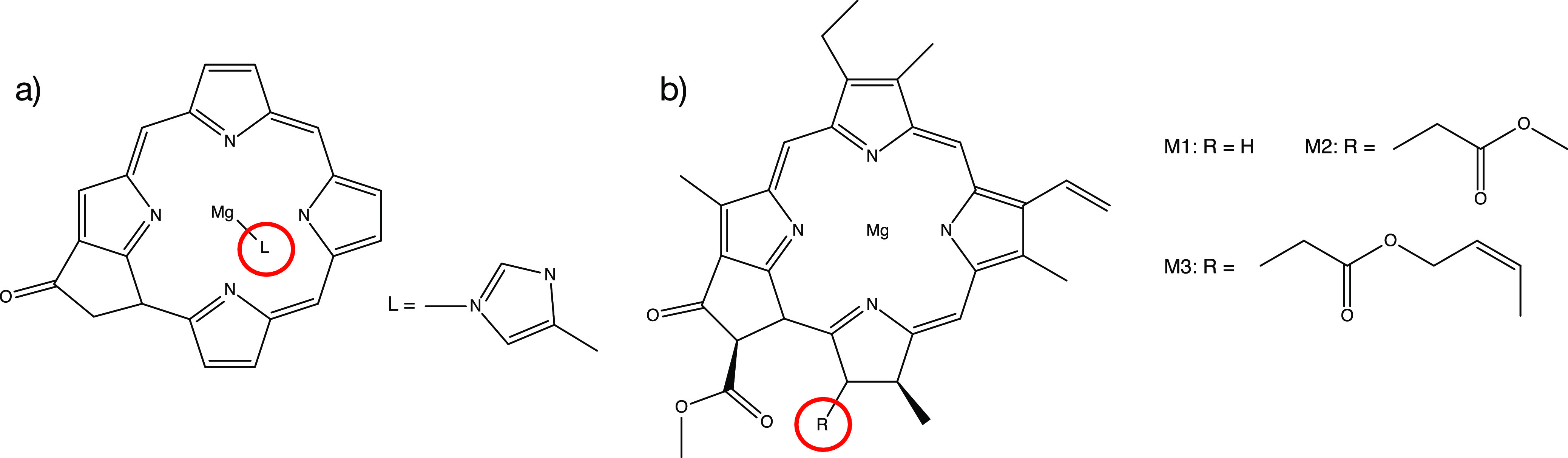
The -Bethe-Salpeter equation (BSE) method is promising for calculating the low-lying excitonic states of molecular systems. However, so far it has only been applied to rather small molecules and in the commonly implemented diagonal approximations to the electronic self-energy, it depends on a mean-field starting point. We describe here an implementation of the self-consistent and starting-point-independent quasiparticle self-consistent (qs)-BSE approach, which is suitable for calculations on large molecules. We herein show that eigenvalue-only self-consistency can lead to an unfaithful description of some excitonic states for chlorophyll dimers while the qs-BSE vertical excitation energies (VEEs) are in excellent agreement with spectroscopic experiments for chlorophyll monomers and dimers measured in the gas phase. Furthermore, VEEs from time-dependent density functional theory calculations tend to disagree with experimental values and using different range-separated hybrid (RSH) kernels does change the VEEs by up to 0.5 eV. We use the new qs-BSE implementation to calculate the lowest excitation energies of the six chromophores of the photosystem II (PSII) reaction center (RC) with nearly 2000 correlated electrons. Using more than 11,000 (6000) basis functions, the calculation could be completed in less than 5 (2) days on a single modern compute node. In agreement with previous TD-DFT calculations using RSH kernels on models that also do not include environmental effects, our qs-BSE calculations only yield states with local characters in the low-energy spectrum of the hexameric complex. Earlier works with RSH kernels have demonstrated that the protein environment facilitates the experimentally observed interchromophoric charge transfer. Therefore, future research will need to combine correlation effects beyond TD-DFT with an explicit treatment of environmental electrostatics.
Bethe-Salpeter 方程 (BSE) 方法在计算分子体系的低能激子态方面很有前景。然而,到目前为止,它只被应用于相当小的分子,并且在通常实现的电子自能对角近似中,它依赖于平均场起点。我们在这里描述了自洽和起点独立的准粒子自洽 (qs)-BSE 方法的实现,该方法适用于大分子的计算。我们在此表明,仅特征值自洽可能导致叶绿素二聚体的一些激子态的描述不真实,而 qs-BSE 垂直激发能 (VEE) 与气相中测量的叶绿素单体和二聚体的光谱实验非常吻合。此外,基于时间相关密度泛函理论的 VEE 计算往往与实验值不一致,并且使用不同的范围分离混合 (RSH) 核可以使 VEE 改变高达 0.5 eV。我们使用新的 qs-BSE 实现来计算光合作用系统 II (PSII) 反应中心 (RC) 的六个发色团的最低激发能,其中包含近 2000 个相关电子。使用超过 11000(6000)个基函数,在单个现代计算节点上不到 5(2)天即可完成计算。与之前使用 RSH 核在不包括环境效应的模型上进行的 TD-DFT 计算一致,我们的 qs-BSE 计算仅在六聚体复合物的低能谱中产生具有局域特征的态。早期使用 RSH 核的工作已经表明,蛋白质环境促进了实验观察到的发色团间电荷转移。因此,未来的研究将需要将超越 TD-DFT 的相关效应与对环境静电的显式处理结合起来。