Department of Biology and Institute for Applied Life Sciences, University of Massachusetts Amherst; Amherst, Massachusetts, United States of America.
Institute for Neurodegenerative Diseases, Weill Institute for Neurosciences, University of California, San Francisco; San Francisco, California, United States of America.
PLoS Pathog. 2022 Dec 1;18(12):e1010956. doi: 10.1371/journal.ppat.1010956. eCollection 2022 Dec.
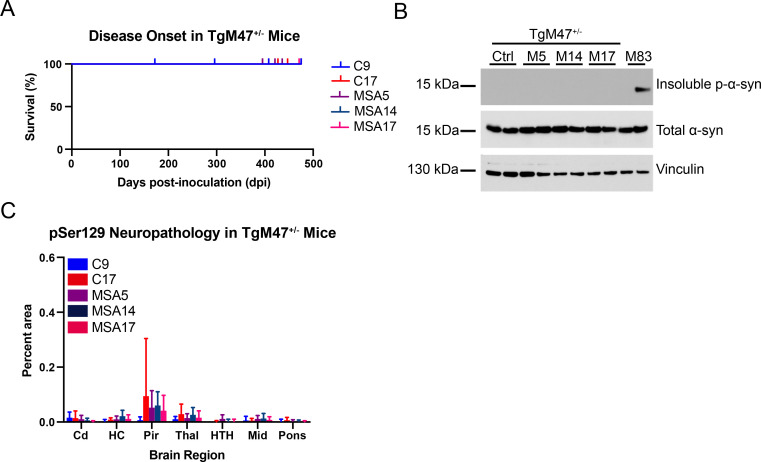
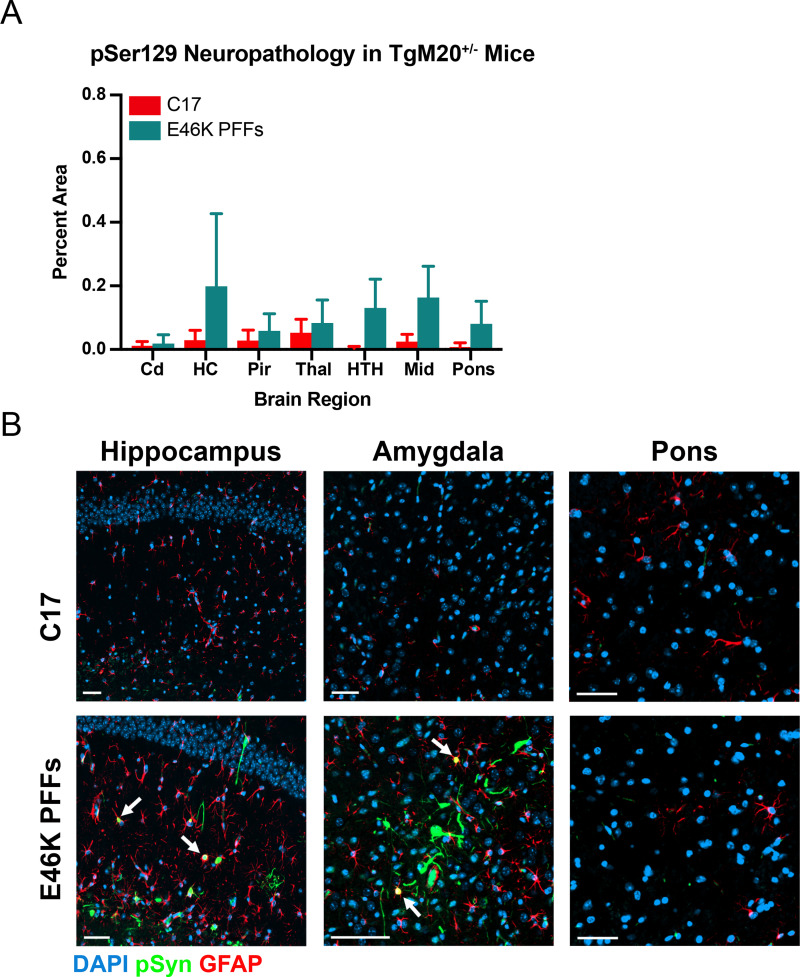
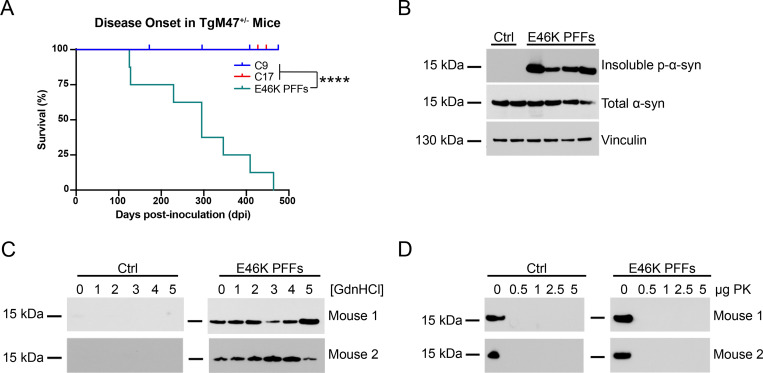
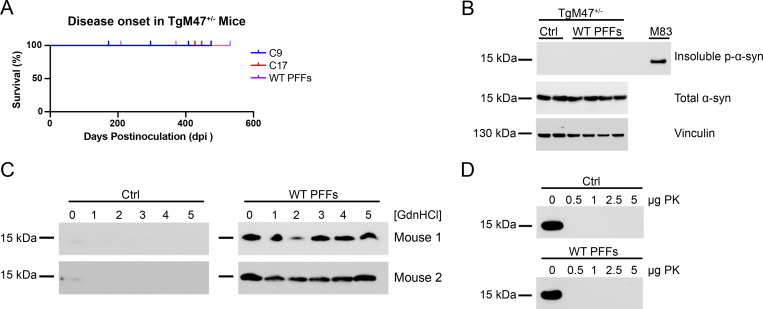
In multiple system atrophy (MSA), the α-synuclein protein misfolds into a self-templating prion conformation that spreads throughout the brain, leading to progressive neurodegeneration. While the E46K mutation in α-synuclein causes familial Parkinson's disease (PD), we previously discovered that this mutation blocks in vitro propagation of MSA prions. Recent studies by others indicate that α-synuclein adopts a misfolded conformation in MSA in which a Greek key motif is stabilized by an intramolecular salt bridge between residues E46 and K80. Hypothesizing that the E46K mutation impedes salt bridge formation and, therefore, exerts a selective pressure that can modulate α-synuclein strain propagation, we asked whether three distinct α-synuclein prion strains could propagate in TgM47+/- mice, which express human α-synuclein with the E46K mutation. Following intracranial injection of these strains, TgM47+/- mice were resistant to MSA prion transmission, whereas recombinant E46K preformed fibrils (PFFs) transmitted neurological disease to mice and induced the formation of phosphorylated α-synuclein neuropathology. In contrast, heterotypic seeding following wild-type (WT) PFF-inoculation resulted in preclinical α-synuclein prion propagation. Moreover, when we inoculated TgM20+/- mice, which express WT human α-synuclein, with E46K PFFs, we observed delayed transmission kinetics with an incomplete attack rate. These findings suggest that the E46K mutation constrains the number of α-synuclein prion conformations that can propagate in TgM47+/- mice, expanding our understanding of the selective pressures that impact α-synuclein prion replication.
在多系统萎缩症(MSA)中,α-突触核蛋白错误折叠成自我模板的朊病毒构象,在大脑中扩散,导致进行性神经退行性变。虽然α-突触核蛋白的 E46K 突变导致家族性帕金森病(PD),但我们之前发现该突变阻止了 MSA 朊病毒的体外传播。最近其他人的研究表明,α-突触核蛋白在 MSA 中采用错误折叠的构象,其中希腊钥匙基序通过 E46 和 K80 残基之间的分子内盐桥稳定。假设 E46K 突变阻碍盐桥形成,因此施加选择性压力,可以调节α-突触核蛋白株的传播,我们询问是否可以在表达 E46K 突变的人类α-突触核蛋白的 TgM47+/- 小鼠中传播三种不同的α-突触核蛋白朊病毒株。在这些菌株的颅内注射后,TgM47+/- 小鼠对 MSA 朊病毒的传播具有抗性,而重组 E46K 预形成纤维(PFF)将神经疾病传播给小鼠,并诱导磷酸化α-突触核蛋白神经病理学的形成。相比之下,在 WT PFF 接种后进行异型播种导致了临床前α-突触核蛋白朊病毒的传播。此外,当我们用 E46K PFF 接种表达 WT 人类α-突触核蛋白的 TgM20+/- 小鼠时,我们观察到传播动力学延迟,且攻击率不完全。这些发现表明,E46K 突变限制了可以在 TgM47+/- 小鼠中传播的α-突触核蛋白朊病毒构象的数量,扩展了我们对影响α-突触核蛋白朊病毒复制的选择性压力的理解。