Department of Biological Physical Chemistry, Rocasolano Institute of Physical Chemistry, CSIC, 28006 Madrid, Spain.
Bioinformatic Unit, Centro de Biología Molecular "Severo Ochoa," CSIC-UAM Cantoblanco, Madrid 28049, Spain.
Bioinformatics. 2023 Jan 1;39(1). doi: 10.1093/bioinformatics/btad011.
Structure-based stability prediction upon mutation is crucial for protein engineering and design, and for understanding genetic diseases or drug resistance events. For this task, we adopted a simple residue-based orientational potential that considers only three backbone atoms, previously applied in protein modeling. Its application to stability prediction only requires parametrizing 12 amino acid-dependent weights using cross-validation strategies on a curated dataset in which we tried to reduce the mutations that belong to protein-protein or protein-ligand interfaces, extreme conditions and the alanine over-representation.
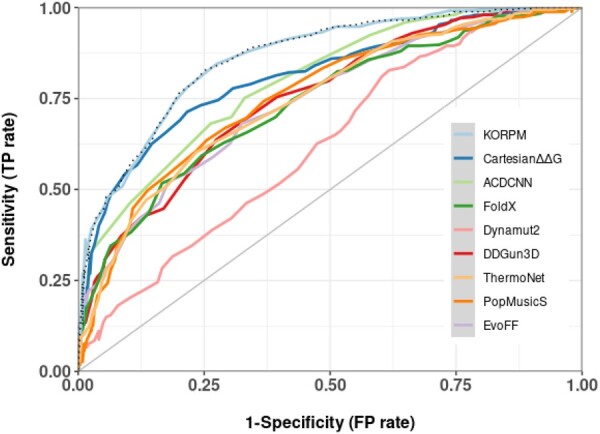
Our method, called KORPM, accurately predicts mutational effects on an independent benchmark dataset, whether the wild-type or mutated structure is used as starting point. Compared with state-of-the-art methods on this balanced dataset, our approach obtained the lowest root mean square error (RMSE) and the highest correlation between predicted and experimental ΔΔG measures, as well as better receiver operating characteristics and precision-recall curves. Our method is almost anti-symmetric by construction, and it performs thus similarly for the direct and reverse mutations with the corresponding wild-type and mutated structures. Despite the strong limitations of the available experimental mutation data in terms of size, variability, and heterogeneity, we show competitive results with a simple sum of energy terms, which is more efficient and less prone to overfitting.
https://github.com/chaconlab/korpm.
Supplementary data are available at Bioinformatics online.
基于结构的突变稳定性预测对于蛋白质工程和设计以及理解遗传疾病或耐药事件至关重要。为此任务,我们采用了一种简单的基于残基的定向势能,仅考虑三个骨干原子,先前已应用于蛋白质建模。其在稳定性预测中的应用仅需要使用交叉验证策略对经过精心整理的数据集进行参数化,我们试图减少属于蛋白质-蛋白质或蛋白质-配体界面、极端条件和丙氨酸过表达的突变。
我们的方法称为 KORPM,可准确预测独立基准数据集上的突变效应,无论是使用野生型还是突变型结构作为起点。与该平衡数据集上的最先进方法相比,我们的方法在预测和实验 ΔΔG 测量之间具有最低的均方根误差 (RMSE) 和最高的相关性,以及更好的接收者操作特征和精度-召回曲线。我们的方法在构建时几乎是反对称的,因此对于具有相应野生型和突变型结构的直接和反向突变,其性能相似。尽管可用的实验突变数据在大小、可变性和异质性方面存在很强的局限性,但我们使用简单的能量项和和具有竞争力的结果,该方法效率更高,不易过拟合。
https://github.com/chaconlab/korpm。
补充数据可在“Bioinformatics”在线获取。