Department of Genetics, School of Biological Sciences, Madurai Kamaraj University, Madurai, 625021, India.
National Institute of Animal Biotechnology, Hyderabad, 500032, India.
BMC Genomics. 2023 Jan 25;24(1):44. doi: 10.1186/s12864-022-09090-7.
Bovine mastitis accounts for significant economic losses to the dairy industry worldwide. Staphylococcus aureus is the most common causative agent of bovine mastitis. Investigating the prevalence of virulence factors and antimicrobial resistance would provide insight into the molecular epidemiology of mastitis-associated S. aureus strains. The present study is focused on the whole genome sequencing and comparative genomic analysis of 41 mastitis-associated S. aureus strains isolated from India.
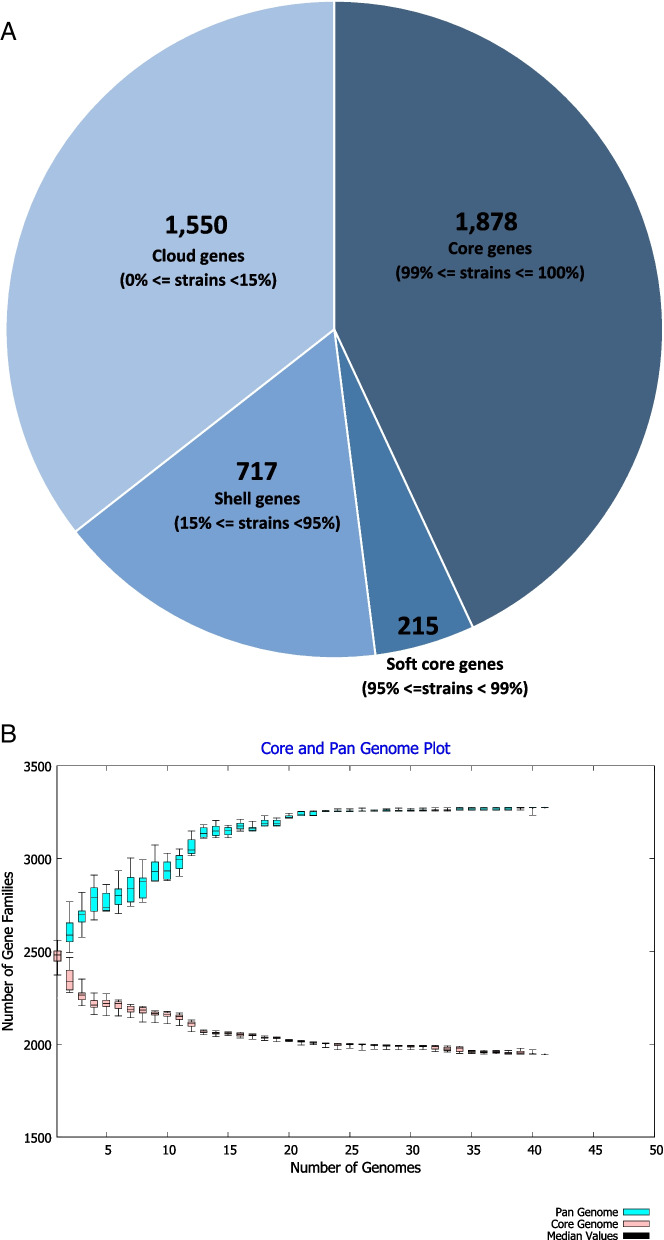
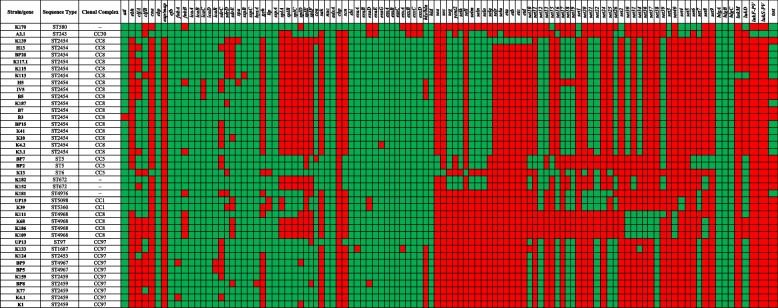
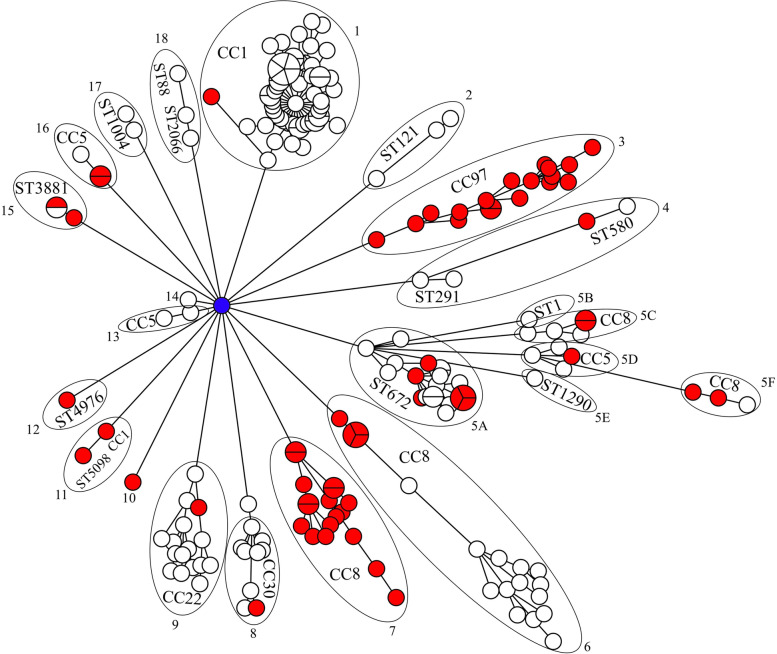
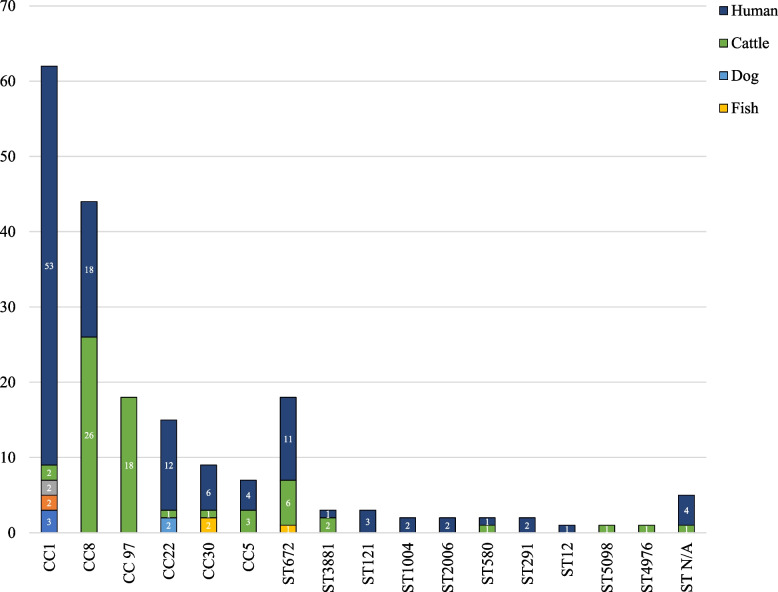
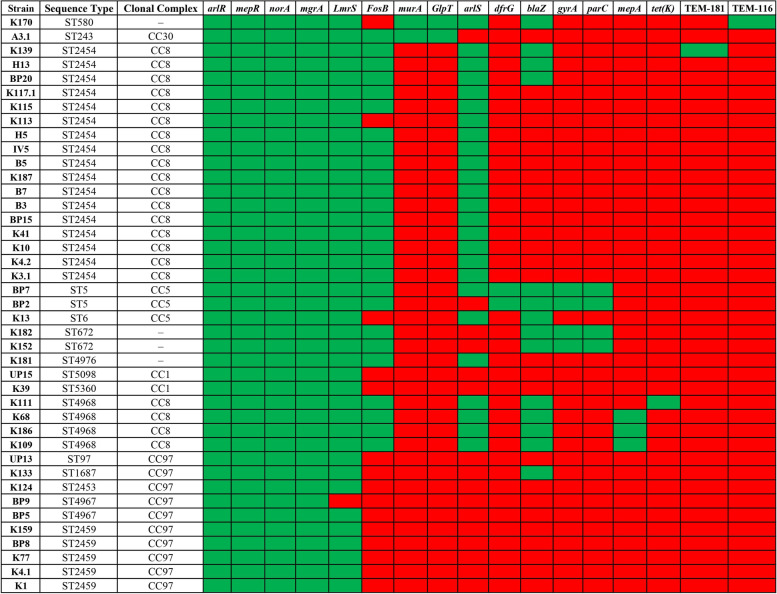
The results elucidate explicit knowledge of 15 diverse sequence types (STs) and five clonal complexes (CCs). The clonal complexes CC8 and CC97 were found to be the predominant genotypes comprising 21 and 10 isolates, respectively. The mean genome size was 2.7 Mbp with a 32.7% average GC content. The pan-genome of the Indian strains of mastitis-associated S. aureus is almost closed. The genome-wide SNP-based phylogenetic analysis differentiated 41 strains into six major clades. Sixteen different spa types were identified, and eight isolates were untypeable. The cgMLST analysis of all S. aureus genome sequences reported from India revealed that S. aureus strain MUF256, isolated from wound fluids of a diabetic patient, was the common ancestor. Further, we observed that all the Indian mastitis-associated S. aureus isolates belonging to the CC97 are mastitis-associated. We identified 17 different antimicrobial resistance (AMR) genes among these isolates, and all the isolates used in this study were susceptible to methicillin. We also identified 108 virulence-associated genes and discuss their associations with different genotypes.
This is the first study presenting a comprehensive whole genome analysis of bovine mastitis-associated S. aureus isolates from India. Comparative genomic analysis revealed the genome diversity, major genotypes, antimicrobial resistome, and virulome of clinical and subclinical mastitis-associated S. aureus strains.
牛乳腺炎在全球范围内给奶业造成了重大的经济损失。金黄色葡萄球菌是引起牛乳腺炎的最常见病原体。研究毒力因子和抗菌药物耐药性的流行情况,可以深入了解乳腺炎相关金黄色葡萄球菌菌株的分子流行病学。本研究集中于对从印度分离的 41 株乳腺炎相关金黄色葡萄球菌进行全基因组测序和比较基因组分析。
结果阐明了 15 种不同序列型(ST)和 5 种克隆群(CC)的明确知识。克隆群 CC8 和 CC97 是主要的基因型,分别包含 21 株和 10 株分离株。平均基因组大小为 2.7 Mbp,平均 GC 含量为 32.7%。乳腺炎相关金黄色葡萄球菌印度株的泛基因组几乎是封闭的。基于全基因组 SNP 的系统发育分析将 41 株菌株分为 6 个主要分支。鉴定出 16 种不同的 spa 型,8 种分离株无法分型。对所有从印度报告的金黄色葡萄球菌基因组序列进行 cgMLST 分析表明,从糖尿病患者伤口液中分离的 MUF256 菌株是金黄色葡萄球菌的共同祖先。此外,我们观察到属于 CC97 的所有印度乳腺炎相关金黄色葡萄球菌分离株均与乳腺炎相关。在这些分离株中,我们鉴定出 17 种不同的抗菌药物耐药(AMR)基因,所有用于本研究的分离株均对甲氧西林敏感。我们还鉴定出 108 种与毒力相关的基因,并讨论了它们与不同基因型的关系。
这是首次对来自印度的牛乳腺炎相关金黄色葡萄球菌分离株进行全面的全基因组分析。比较基因组分析揭示了乳腺炎相关金黄色葡萄球菌临床和亚临床分离株的基因组多样性、主要基因型、抗菌药物耐药组和毒力组。