College of Forestry and Landscape Architecture, South China Agricultural University, Guangzhou, 51000, Guangdong, China.
Sichuan Academy of Forestry Sciences, Chengdu, 61008, Sichuan, China.
BMC Genomics. 2023 Feb 1;24(1):58. doi: 10.1186/s12864-023-09150-6.
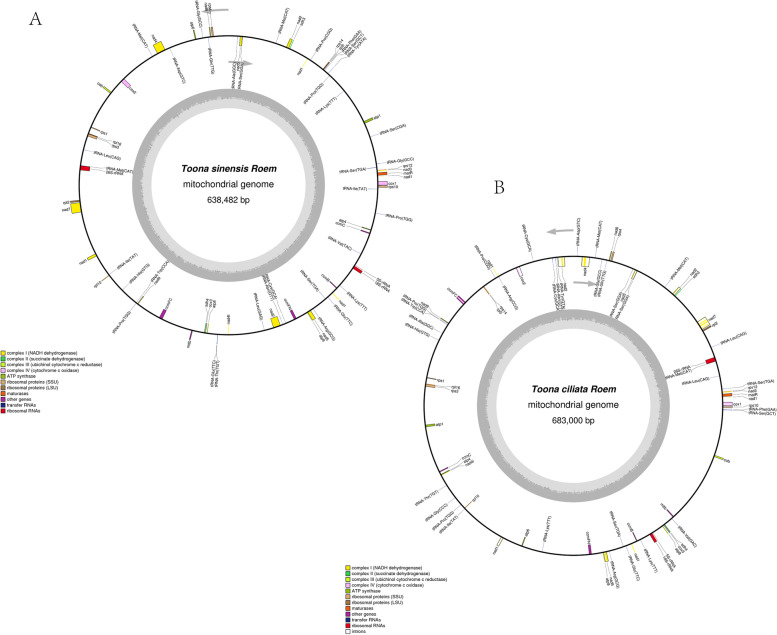
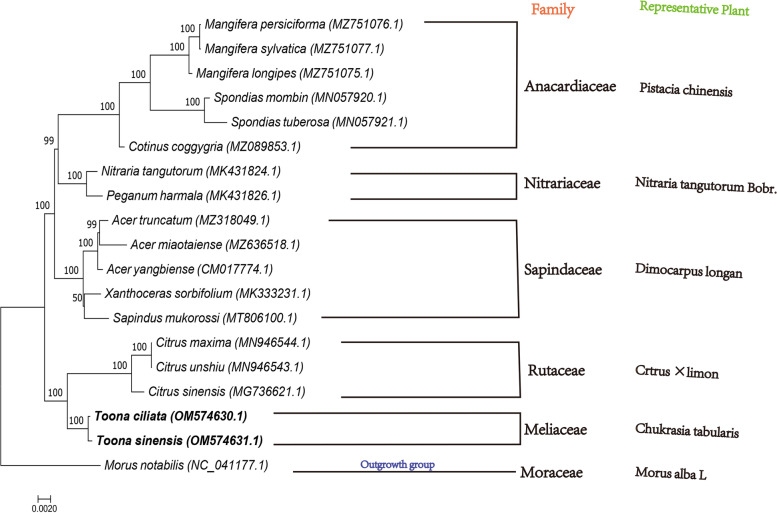
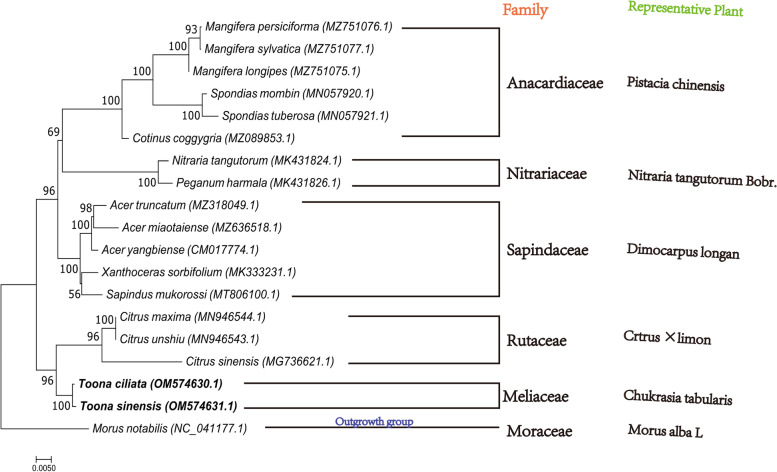
Toona is a critical genus in the Meliaceae, and the plants of this group are an asset for both restorative and restorative purposes, the most flexible of which are Toona sinensis and Toona ciliata. To concentrate on the advancement of mitochondrial(Mt) genome variety in T.sinensis and T.ciliata, the Mt genomes of the two species were sequenced in high throughput independently, after de novo assembly and annotation to construct a Mt genome map for comparison in genome structure. Find their repetitive sequences and analyze them in comparison with the chloroplast genome, along with Maximum-likelihood(ML) phylogenetic analysis with 16 other relatives.
(1) T. sinensis and T.ciliata are both circular structures with lengths of 683482 bp and 68300 bp, respectively. They share a high degree of similarity in encoding genes and have AT preferences. All of them have the largest Phe concentration and are the most frequently used codons. (2) Both of their Mt genome are highly preserved in terms of structural and functional genes, while the main variability is reflected in the length of tRNA, the number of genes, and the value of RSCU. (3) T. siniensis and T. ciliata were detected to have 94 and 87 SSRs, respectively, of which mononucleotides accounted for the absolute proportion. Besides, the vast majority of their SSRs were found to be poly-A or poly-T. (4)10 and 11 migrating fragments were identified in the comparison with the chloroplast genome, respectively. (5) In the ML evolutionary tree, T.sinensis and T.ciliata clustered individually into a small branch with 100% support, reflecting two species of Toona are very similarly related to each other.
This research provides a basis for the exploitation of T.sinensis and T.ciliata in terms of medicinal, edible, and timber resources to avoid confusion; at the same time, it can explore the evolutionary relationship between the Toona and related species, which does not only have an important practical value, but also provides a theoretical basis for future hybrid breeding of forest trees, molecular markers, and evolutionary aspects of plants, which has great scientific significance.
香椿是楝科的一个关键属,该类植物具有药用和食用价值,其中最灵活的是香椿和红椿。为了集中研究香椿和红椿的线粒体(Mt)基因组多样性,我们独立地对这两个物种的 Mt 基因组进行了高通量测序,然后进行从头组装和注释,以构建 Mt 基因组图谱进行基因组结构比较。我们发现了它们的重复序列,并与叶绿体基因组进行了比较,同时还进行了包括 16 个其他亲缘关系的最大似然(ML)系统发育分析。
(1)香椿和红椿都是圆形结构,长度分别为 683482bp 和 68300bp。它们在编码基因方面具有高度的相似性,并且偏好使用 AT。它们都具有最大的 Phe 浓度,并且是最常用的密码子。(2)它们的 Mt 基因组在结构和功能基因方面都高度保守,而主要的变异性反映在 tRNA 的长度、基因的数量和 RSCU 值上。(3)香椿和红椿分别检测到 94 和 87 个 SSRs,其中单核苷酸占绝对比例。此外,它们的 SSR 绝大多数是 poly-A 或 poly-T。(4)与叶绿体基因组比较,分别鉴定出 10 和 11 个迁移片段。(5)在 ML 进化树中,香椿和红椿分别单独聚类成一个小分支,支持率为 100%,反映出两种香椿的亲缘关系非常相似。
本研究为香椿和红椿的药用、食用和木材资源开发提供了基础,避免了混淆;同时,它还可以探索香椿与相关物种的进化关系,不仅具有重要的实用价值,而且为未来的林木杂交育种、分子标记和植物进化等方面提供了理论基础,具有重要的科学意义。