Chen Jingxuan, Basting Preston J, Han Shunhua, Garfinkel David J, Bergman Casey M
Institute of Bioinformatics, University of Georgia, Athens, GA.
Department of Biochemistry and Molecular Biology, University of Georgia, Athens, GA.
bioRxiv. 2023 Mar 21:2023.02.13.528343. doi: 10.1101/2023.02.13.528343.
Many computational methods have been developed to detect non-reference transposable element (TE) insertions using short-read whole genome sequencing data. The diversity and complexity of such methods often present challenges to new users seeking to reproducibly install, execute, or evaluate multiple TE insertion detectors.
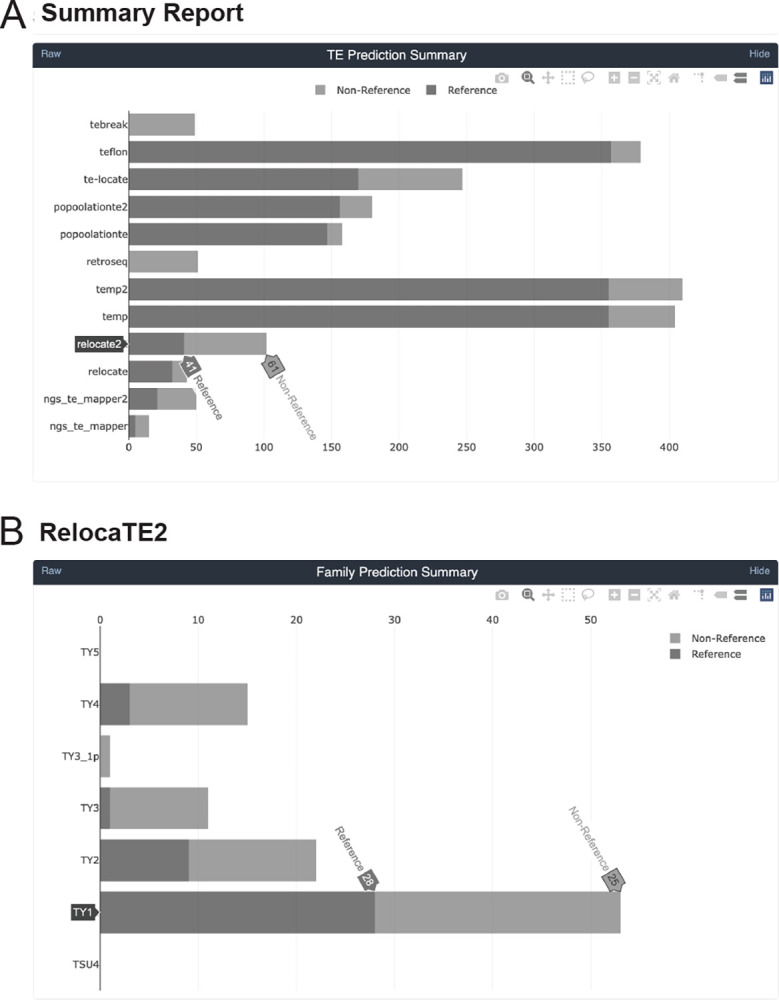
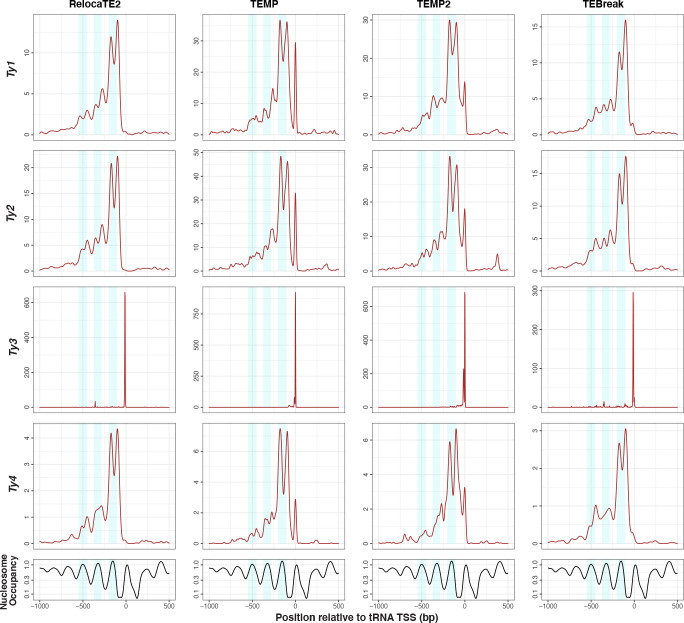
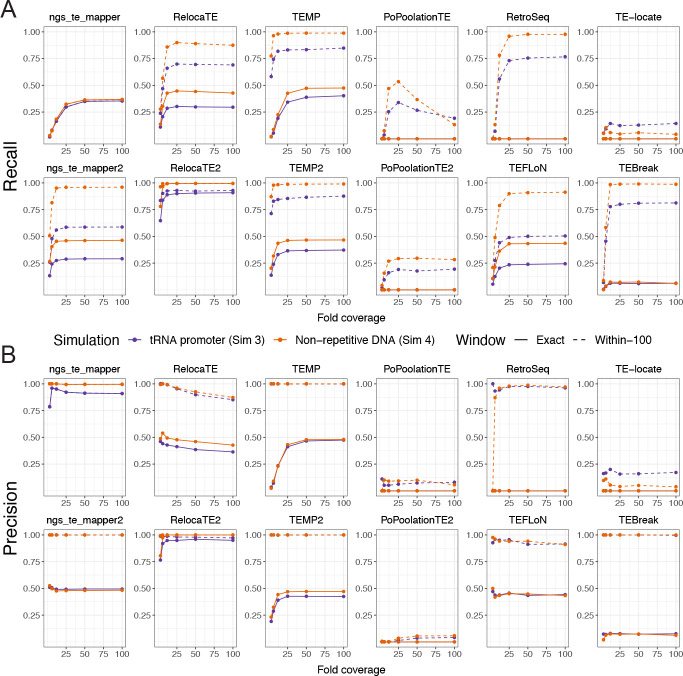
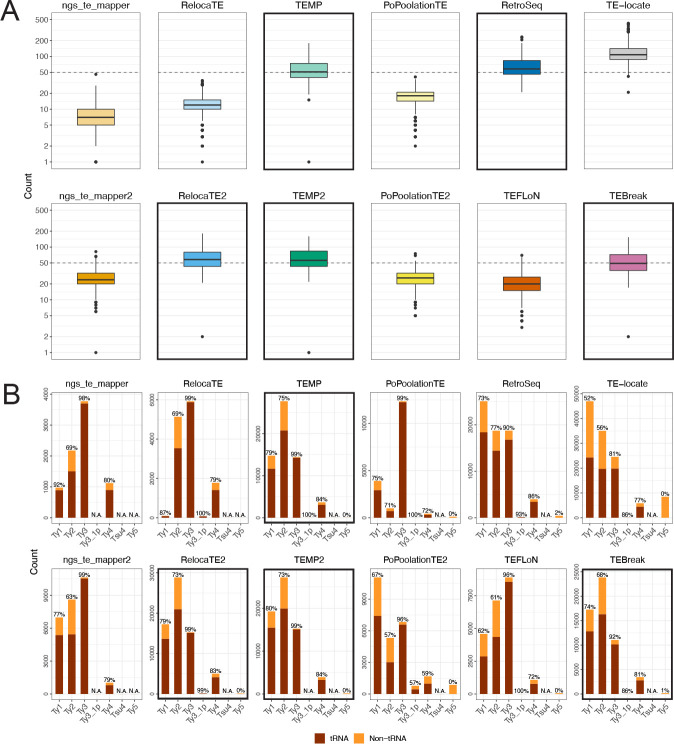
We previously developed the McClintock meta-pipeline to facilitate the installation, execution, and evaluation of six first-generation short-read TE detectors. Here, we report a completely re-implemented version of McClintock written in Python using Snakemake and Conda that improves its installation, error handling, speed, stability, and extensibility. McClintock 2 now includes 12 short-read TE detectors, auxiliary pre-processing and analysis modules, interactive HTML reports, and a simulation framework to reproducibly evaluate the accuracy of component TE detectors. When applied to the model microbial eukaryote Saccharomyces cerevisiae, we find substantial variation in the ability of McClintock 2 components to identify the precise locations of non-reference TE insertions, with RelocaTE2 showing the highest recall and precision in simulated data. We find that RelocaTE2, TEMP, TEMP2 and TEBreak provide a consistent and biologically meaningful view of non-reference TE insertions in a species-wide panel of ∼1000 yeast genomes, as evaluated by coverage-based abundance estimates and expected patterns of tRNA promoter targeting. Finally, we show that best-in-class predictors for yeast have sufficient resolution to reveal a dyad pattern of integration in nucleosome-bound regions upstream of yeast tRNA genes for Ty1, Ty2, and Ty4, allowing us to extend knowledge about fine-scale target preferences first revealed experimentally for Ty1 to natural insertions and related copia-superfamily retrotransposons in yeast.
McClintock (https://github.com/bergmanlab/mcclintock/) provides a user-friendly pipeline for the identification of TEs in short-read WGS data using multiple TE detectors, which should benefit researchers studying TE insertion variation in a wide range of different organisms. Application of the improved McClintock system to simulated and empirical yeast genome data reveals best-in-class methods and novel biological insights for one of the most widely-studied model eukaryotes and provides a paradigm for evaluating and selecting non-reference TE detectors for other species.
已经开发了许多计算方法,用于使用短读长全基因组测序数据检测非参考转座元件(TE)插入。这些方法的多样性和复杂性常常给试图可重复地安装、执行或评估多个TE插入检测器的新用户带来挑战。
我们之前开发了McClintock元管道,以方便六个第一代短读长TE检测器的安装、执行和评估。在此,我们报告了一个使用Snakemake和Conda以Python编写的完全重新实现的McClintock版本,它改进了安装、错误处理、速度、稳定性和可扩展性。McClintock 2现在包括12个短读长TE检测器、辅助预处理和分析模块、交互式HTML报告以及一个模拟框架,用于可重复地评估组件TE检测器的准确性。当应用于模式微生物真核生物酿酒酵母时,我们发现McClintock 2组件识别非参考TE插入精确位置的能力存在很大差异,RelocaTE2在模拟数据中显示出最高的召回率和精确率。我们发现,通过基于覆盖度的丰度估计和tRNA启动子靶向的预期模式评估,RelocaTE2、TEMP、TEMP2和TEBreak在约1000个酵母基因组的全物种面板中提供了关于非参考TE插入的一致且具有生物学意义的视图。最后,我们表明,酵母中的一流预测器具有足够的分辨率,可揭示酵母tRNA基因上游核小体结合区域中Ty1、Ty2和Ty4的二元整合模式,这使我们能够将最初通过实验揭示的关于Ty1的精细尺度靶标偏好的知识扩展到酵母中的自然插入和相关的copia超家族逆转座子。