Aziz Shahid, Rasheed Faisal, Zahra Rabaab, König Simone
Patients Diagnostic Lab, Pakistan Institute of Nuclear Science and Technology (PINSTEC), Islamabad 44000, Pakistan.
Department of Microbiology, Faculty of Biological Sciences, Quaid-i-Azam University, Islamabad 45320, Pakistan.
Life (Basel). 2023 Feb 15;13(2):544. doi: 10.3390/life13020544.
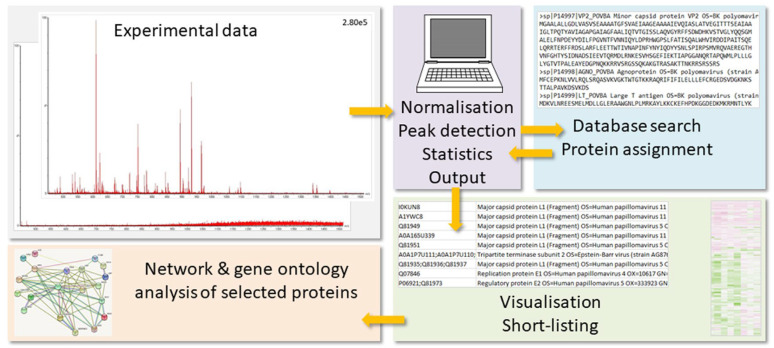
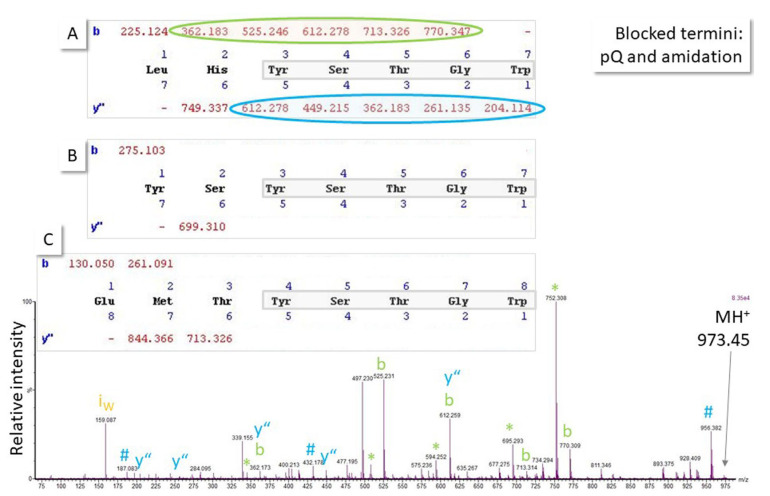
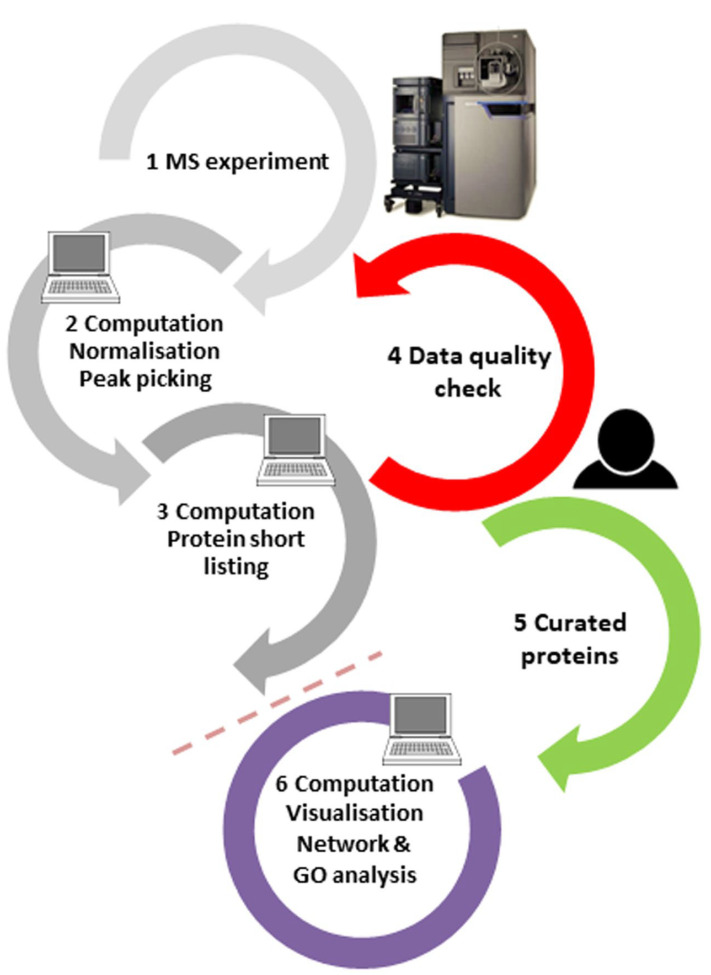
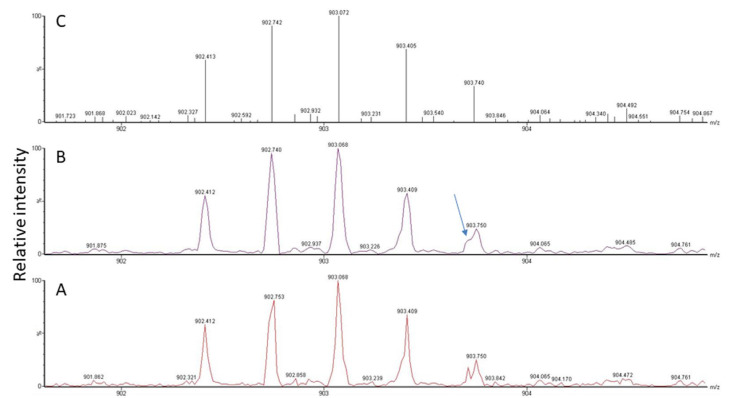
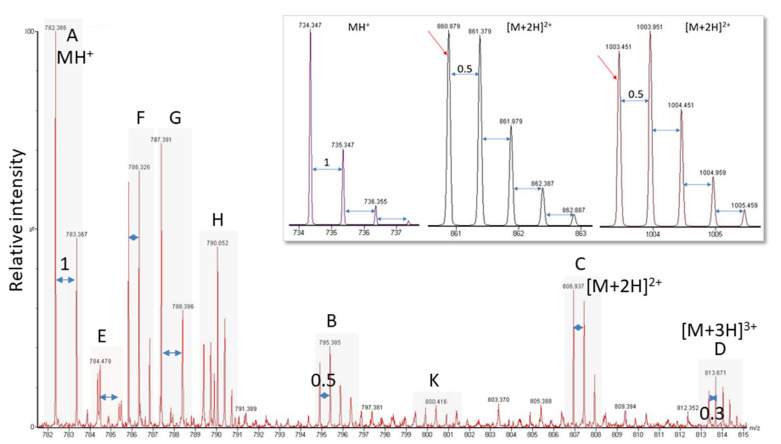
(1) Background: Untargeted mass spectrometry (MS)-based proteomic analysis is highly amenable to automation. Software algorithms translate raw spectral data into protein information obtained by a comparison to sequence databases. However, the technology has limitations, especially for analytes measured at the limit of detection. In a protein expression study of human gastric biopsies, the question arose whether or not it is possible, as well as sensible, to search for viral proteins in addition to those from the human host. (2) Methods: Experimental data-independent MS data were analyzed using protein sequences for oncoviruses, and BLAST analyses were performed to elucidate the level of sequence homology to host proteins. (3) Results: About one hundred viral proteins were assigned, but there was also up to 43% sequence homology to human proteins. (4) Conclusions: There are at least two reasons why the matches to viral proteins should be used with care. First, it is not plausible that large amounts of viral proteins should be present in human gastric biopsies, so the spectral quality of the peptides derived from viral proteins is likely low. As a consequence, the number of false assignments is high. Second, homologous peptides found both in human and virus proteomes contribute to matching errors. Thus, though shotgun proteomics raw data can technically be analyzed using any database, meaningful results cannot be always expected and a sanity check must be performed. Both instrumentation and bioinformatic processing in MS-based proteomics are continuously improving at lowering the limit of detection even further. Nevertheless, data output should always be controlled in order to avoid the over-interpretation of results.
(1) 背景:基于非靶向质谱(MS)的蛋白质组学分析非常适合自动化操作。软件算法通过与序列数据库进行比对,将原始光谱数据转化为蛋白质信息。然而,该技术存在局限性,尤其是对于在检测限水平测量的分析物。在一项人类胃活检组织的蛋白质表达研究中,出现了一个问题,即除了人类宿主的蛋白质外,是否有可能以及是否合理去搜索病毒蛋白。(2) 方法:使用肿瘤病毒的蛋白质序列对实验性数据非依赖型MS数据进行分析,并进行BLAST分析以阐明与宿主蛋白的序列同源性水平。(3) 结果:鉴定出约100种病毒蛋白,但与人类蛋白的序列同源性高达43%。(4) 结论:对于病毒蛋白匹配结果的使用应谨慎,至少有两个原因。其一,人类胃活检组织中存在大量病毒蛋白这种情况不太合理,所以源自病毒蛋白的肽段的光谱质量可能较低。因此,错误鉴定的数量较多。其二,在人类和病毒蛋白质组中都发现的同源肽段会导致匹配错误。所以,尽管鸟枪法蛋白质组学原始数据在技术上可以使用任何数据库进行分析,但不一定总能得到有意义的结果,必须进行合理性检查。基于MS的蛋白质组学中的仪器设备和生物信息处理都在不断改进,以进一步降低检测限。然而,数据输出始终应受到控制以避免对结果的过度解读。