Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK.
Cavendish Laboratory, Department of Physics, University of Cambridge, Cambridge CB3 0HE, UK.
Proc Natl Acad Sci U S A. 2023 Mar 14;120(11):e2214168120. doi: 10.1073/pnas.2214168120. Epub 2023 Mar 6.
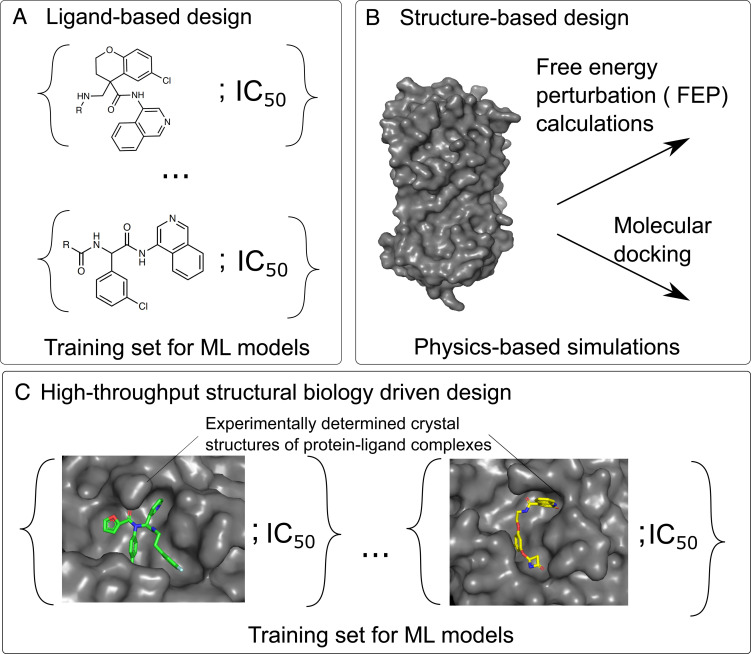
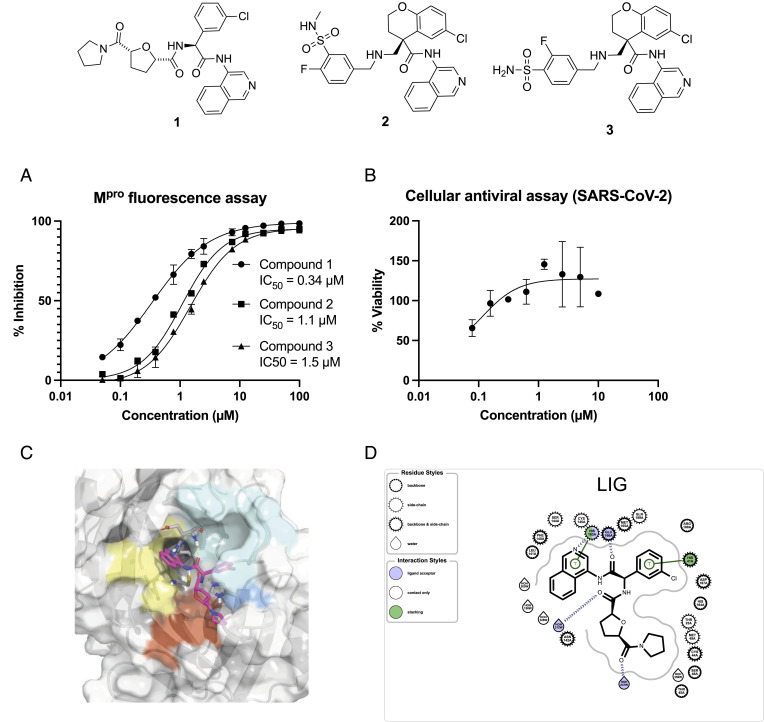
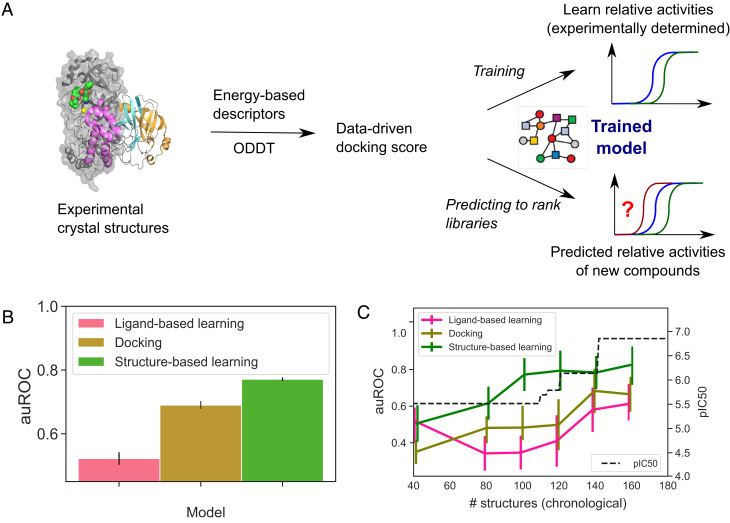
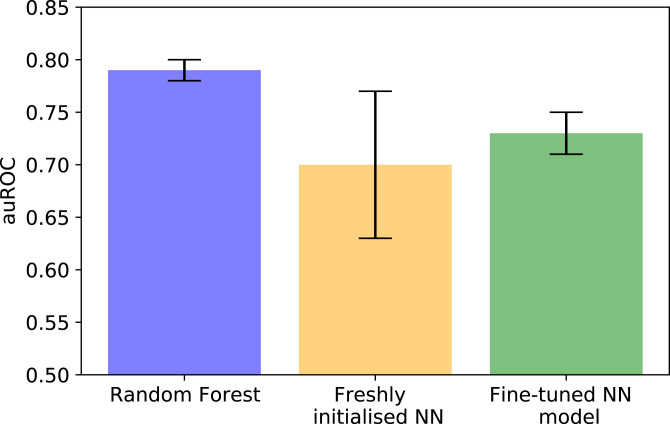
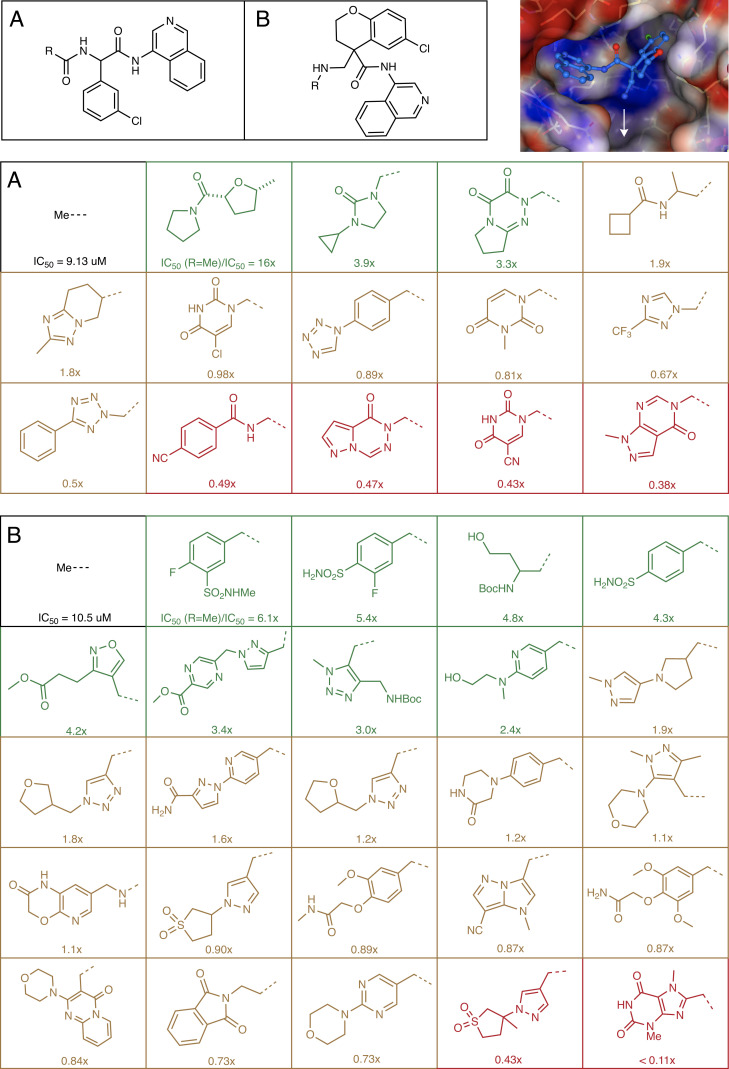
A common challenge in drug design pertains to finding chemical modifications to a ligand that increases its affinity to the target protein. An underutilized advance is the increase in structural biology throughput, which has progressed from an artisanal endeavor to a monthly throughput of hundreds of different ligands against a protein in modern synchrotrons. However, the missing piece is a framework that turns high-throughput crystallography data into predictive models for ligand design. Here, we designed a simple machine learning approach that predicts protein-ligand affinity from experimental structures of diverse ligands against a single protein paired with biochemical measurements. Our key insight is using physics-based energy descriptors to represent protein-ligand complexes and a learning-to-rank approach that infers the relevant differences between binding modes. We ran a high-throughput crystallography campaign against the SARS-CoV-2 main protease (M), obtaining parallel measurements of over 200 protein-ligand complexes and their binding activities. This allows us to design one-step library syntheses which improved the potency of two distinct micromolar hits by over 10-fold, arriving at a noncovalent and nonpeptidomimetic inhibitor with 120 nM antiviral efficacy. Crucially, our approach successfully extends ligands to unexplored regions of the binding pocket, executing large and fruitful moves in chemical space with simple chemistry.
药物设计中一个常见的挑战是找到增加配体与靶蛋白亲和力的化学修饰。一个未被充分利用的进展是结构生物学通量的增加,这已经从一项手工工艺发展到了现代同步加速器中每月对数百种不同配体与一种蛋白质进行高通量筛选。然而,缺少的是一个将高通量晶体学数据转化为配体设计预测模型的框架。在这里,我们设计了一种简单的机器学习方法,可以根据单一蛋白质的不同配体的实验结构和生化测量值预测蛋白质-配体亲和力。我们的关键见解是使用基于物理的能量描述符来表示蛋白质-配体复合物,并使用学习排序方法推断结合模式之间的相关差异。我们针对 SARS-CoV-2 主蛋白酶 (M) 进行了高通量晶体学筛选,获得了超过 200 个蛋白质-配体复合物及其结合活性的平行测量值。这使我们能够设计一步文库合成,将两种不同的微摩尔级命中物的效力提高了 10 倍以上,得到了一种具有 120 nM 抗病毒功效的非共价和非肽模拟抑制剂。至关重要的是,我们的方法成功地将配体扩展到了结合口袋的未探索区域,用简单的化学方法在化学空间中进行了大规模和富有成效的移动。