German National Reference Centre for Borrelia, Oberschleissheim, Germany.
Bavarian Health and Food Safety Authority, Oberschleissheim, Germany.
BMC Genomics. 2023 Jul 17;24(1):401. doi: 10.1186/s12864-023-09500-4.
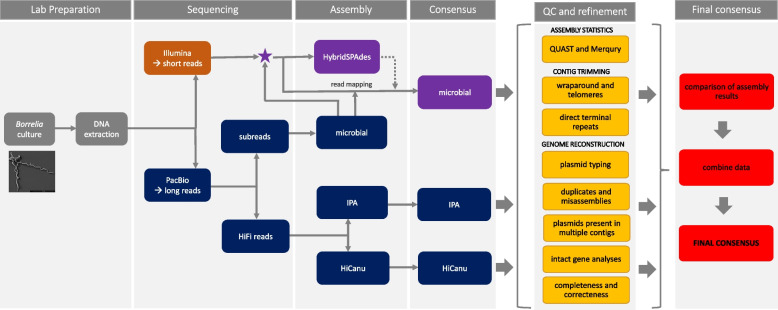
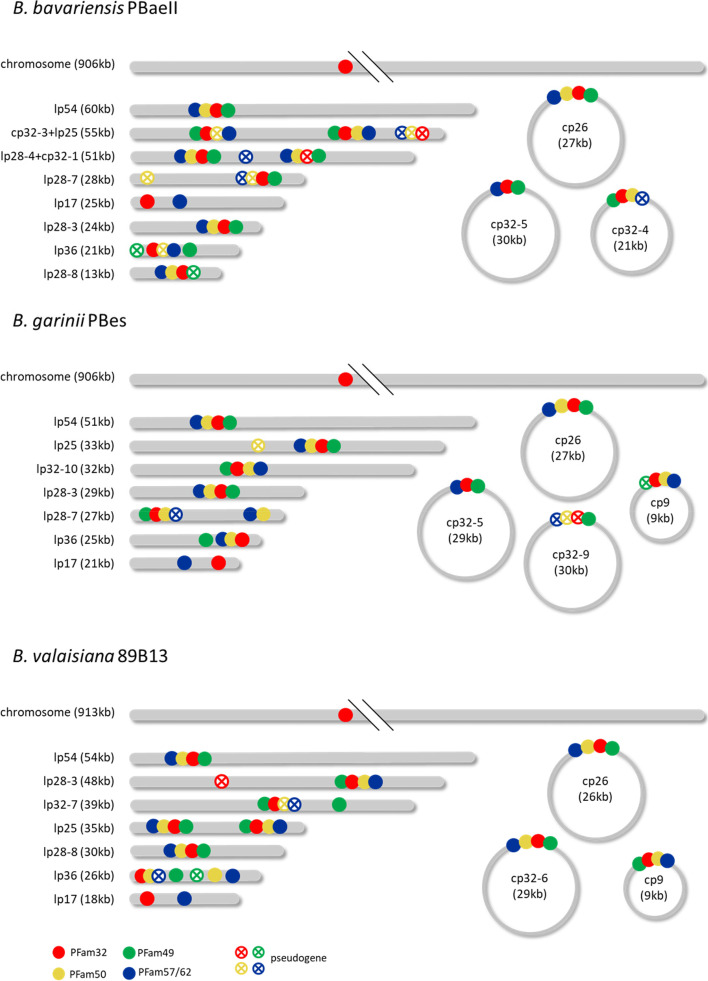
Bacteria of the Borrelia burgdorferi sensu lato (s.l.) complex can cause Lyme borreliosis. Different B. burgdorferi s.l. genospecies vary in their host and vector associations and human pathogenicity but the genetic basis for these adaptations is unresolved and requires completed and reliable genomes for comparative analyses. The de novo assembly of a complete Borrelia genome is challenging due to the high levels of complexity, represented by a high number of circular and linear plasmids that are dynamic, showing mosaic structure and sequence homology. Previous work demonstrated that even advanced approaches, such as a combination of short-read and long-read data, might lead to incomplete plasmid reconstruction. Here, using recently developed high-fidelity (HiFi) PacBio sequencing, we explored strategies to obtain gap-free, complete and high quality Borrelia genome assemblies. Optimizing genome assembly, quality control and refinement steps, we critically appraised existing techniques to create a workflow that lead to improved genome reconstruction.
Despite the latest available technologies, stand-alone sequencing and assembly methods are insufficient for the generation of complete and high quality Borrelia genome assemblies. We developed a workflow pipeline for the de novo genome assembly for Borrelia using several types of sequence data and incorporating multiple assemblers to recover the complete genome including both circular and linear plasmid sequences.
Our study demonstrates that, with HiFi data and an ensemble reconstruction pipeline with refinement steps, chromosomal and plasmid sequences can be fully resolved, even for complex genomes such as Borrelia. The presented pipeline may be of interest for the assembly of further complex microbial genomes.
伯氏疏螺旋体复合种(s.l.)中的细菌可引起莱姆病。不同的伯氏疏螺旋体复合种在宿主和媒介的关联性以及对人类的致病性方面存在差异,但这些适应性的遗传基础尚未确定,需要完成和可靠的基因组进行比较分析。由于存在大量的圆形和线性质粒,其结构复杂且具有动态性、呈现镶嵌结构和序列同源性,因此从头组装完整的伯氏疏螺旋体基因组具有挑战性。先前的研究表明,即使是先进的方法,如短读长和长读长数据的结合,也可能导致质粒的不完全重建。在这里,我们使用最近开发的高保真(HiFi)PacBio 测序技术,探索了获得无缺口、完整和高质量伯氏疏螺旋体基因组组装的策略。通过优化基因组组装、质量控制和细化步骤,我们批判性地评估了现有的技术,以创建一个导致改进基因组重建的工作流程。
尽管有最新的可用技术,但独立的测序和组装方法不足以生成完整和高质量的伯氏疏螺旋体基因组组装。我们开发了一种使用多种类型的序列数据和整合多个组装程序的从头组装伯氏疏螺旋体基因组的工作流程,以恢复包括圆形和线性质粒序列在内的完整基因组。
我们的研究表明,使用 HiFi 数据和具有细化步骤的集成重建管道,可以完全解析染色体和质粒序列,即使对于复杂的基因组,如伯氏疏螺旋体。所提出的工作流程可能对进一步复杂微生物基因组的组装感兴趣。