Institute of Pharmacology and Toxicology, University of Zurich, 8057 Zurich, Switzerland.
Neuroscience Center Zurich, University of Zurich and ETH Zurich, 8057 Zurich, Switzerland.
Int J Mol Sci. 2023 Aug 30;24(17):13436. doi: 10.3390/ijms241713436.
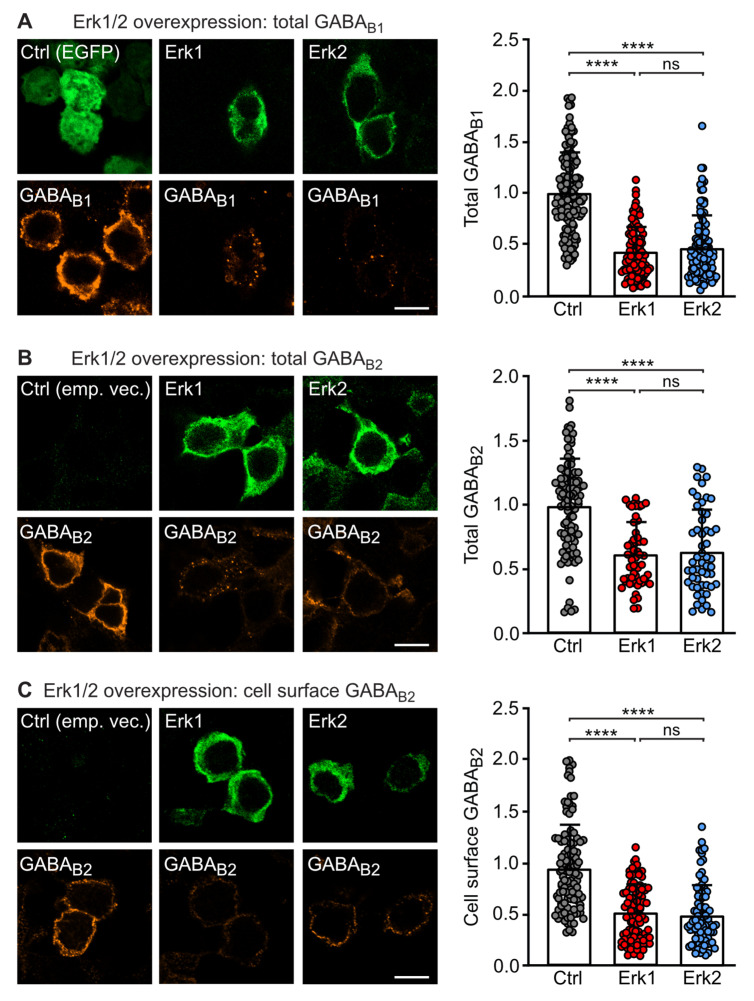
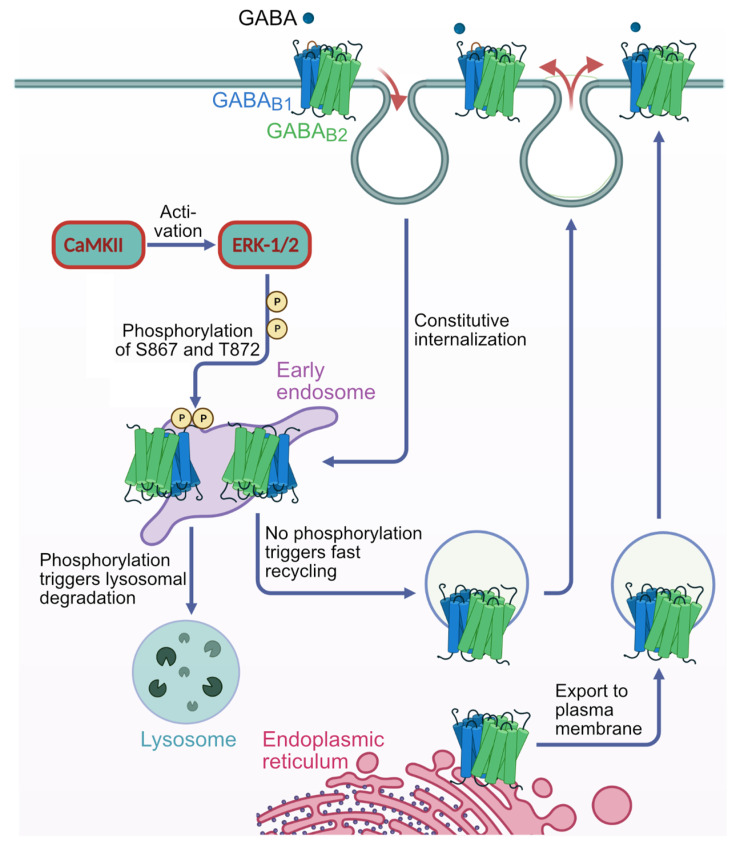
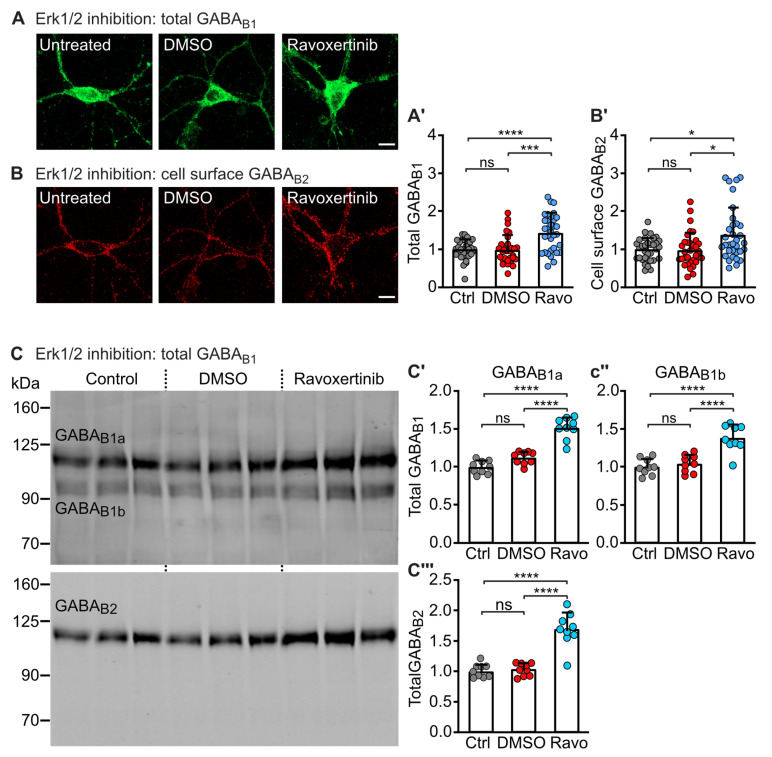
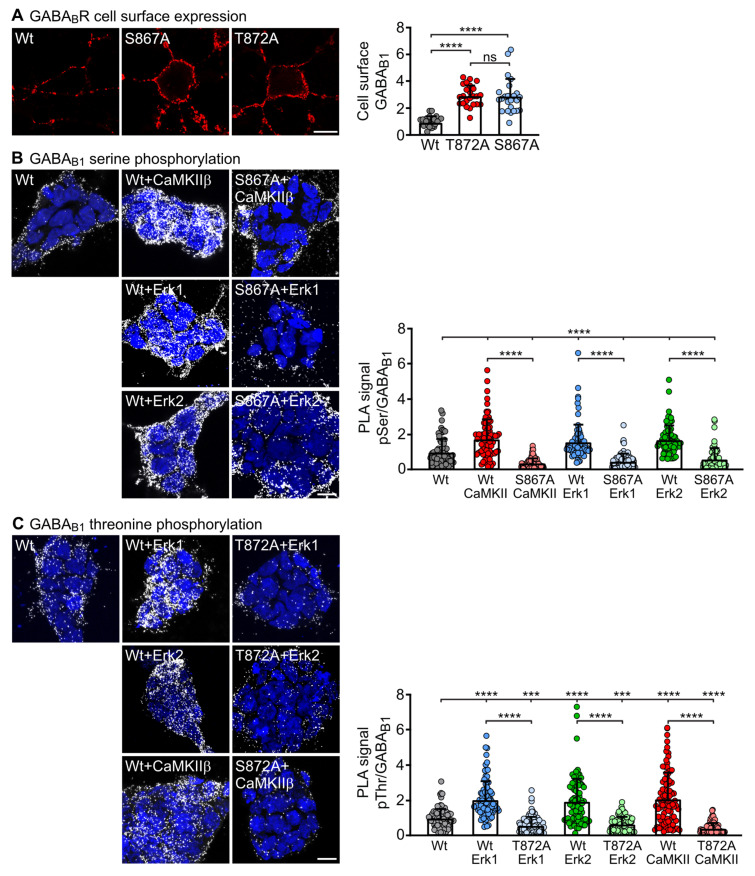
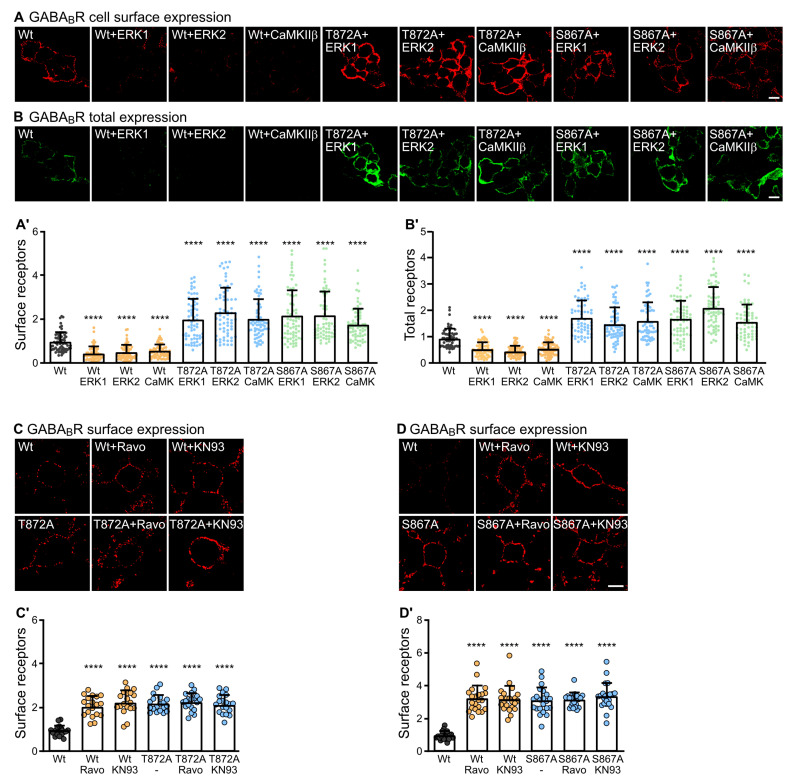
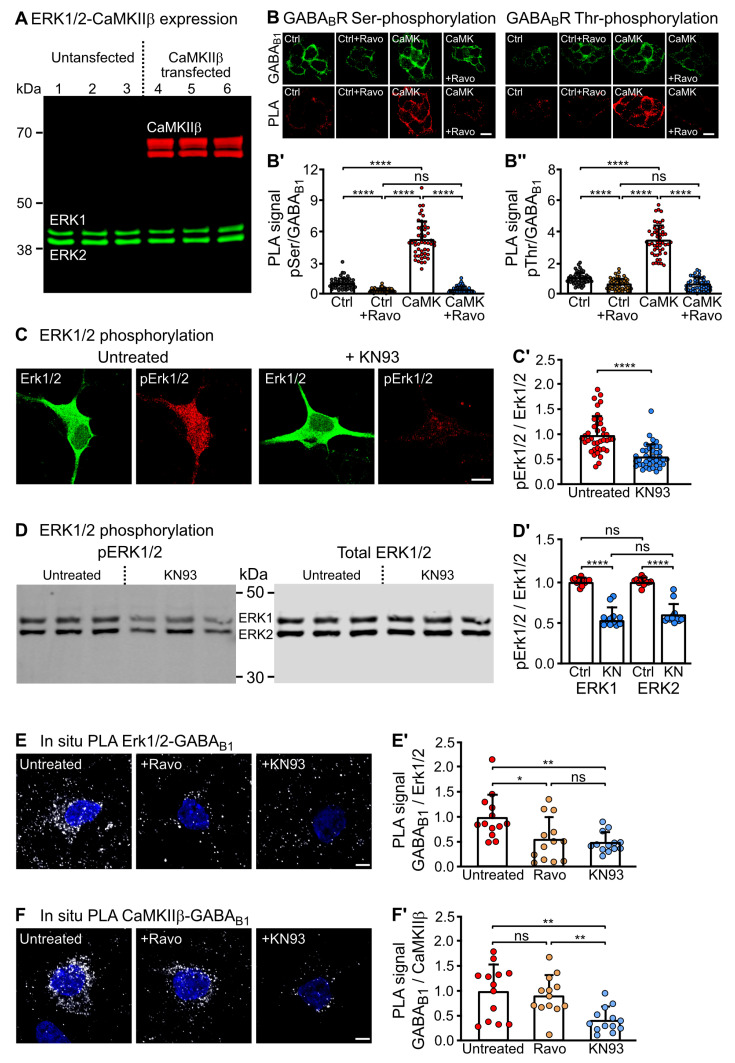
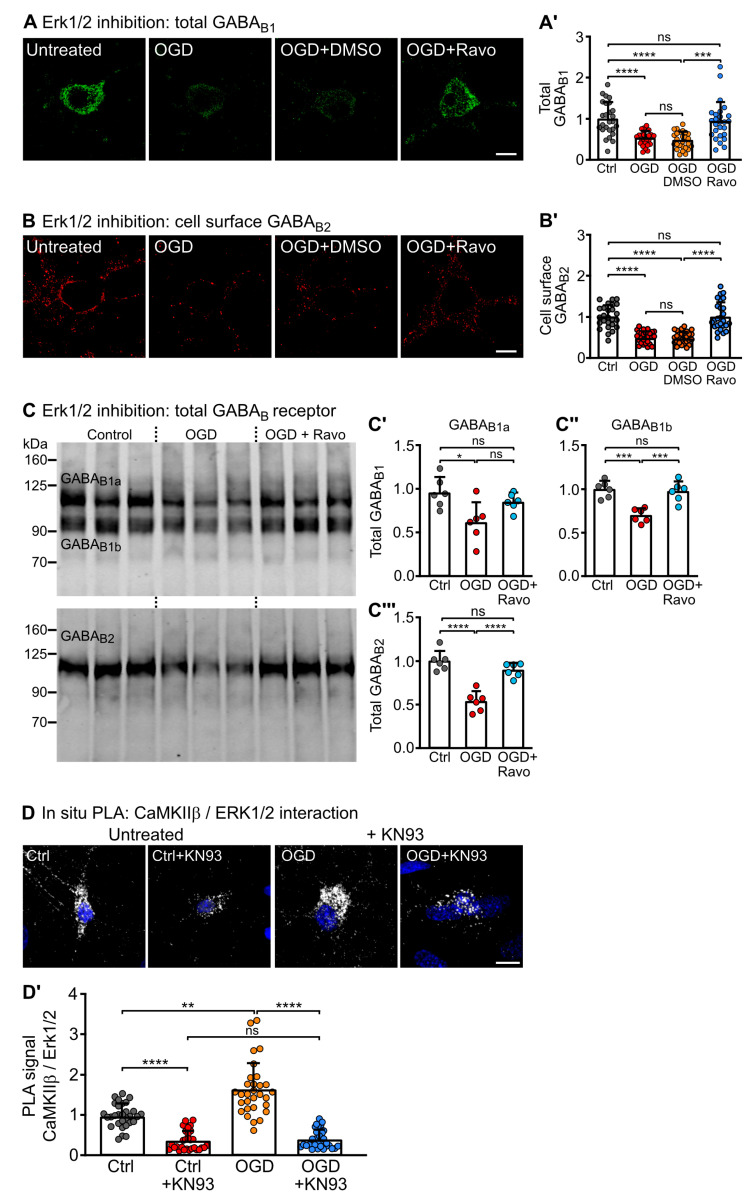
GABA receptor-mediated inhibition is indispensable for maintaining a healthy neuronal excitation/inhibition balance. Many neurological diseases are associated with a disturbed excitation/inhibition balance and downregulation of GABA receptors due to enhanced sorting of the receptors to lysosomal degradation. A key event triggering the downregulation of the receptors is the phosphorylation of S867 in the GABA subunit mediated by CaMKIIβ. Interestingly, close to S867 in GABA exists another phosphorylation site, T872. Therefore, the question arose as to whether phosphorylation of T872 is involved in downregulating the receptors and whether phosphorylation of this site is also mediated by CaMKIIβ or by another protein kinase. Here, we show that mutational inactivation of T872 in GABA prevented the degradation of the receptors in cultured neurons. We found that, in addition to CaMKIIβ, also ERK1/2 is involved in the degradation pathway of GABA receptors under physiological and ischemic conditions. In contrast to our previous view, CaMKIIβ does not appear to directly phosphorylate S867. Instead, the data support a mechanism in which CaMKIIβ activates ERK1/2, which then phosphorylates S867 and T872 in GABA. Blocking ERK activity after subjecting neurons to ischemic stress completely restored downregulated GABA receptor expression to normal levels. Thus, preventing ERK1/2-mediated phosphorylation of S867/T872 in GABA is an opportunity to inhibit the pathological downregulation of the receptors after ischemic stress and is expected to restore a healthy neuronal excitation/inhibition balance.
GABA 受体介导的抑制对于维持健康的神经元兴奋/抑制平衡是必不可少的。许多神经疾病都与兴奋/抑制平衡失调有关,并且由于 GABA 受体向溶酶体降解的增强而导致 GABA 受体的下调。触发受体下调的关键事件是 CaMKIIβ 介导的 GABA 亚基中 S867 的磷酸化。有趣的是,在 GABA 附近存在另一个磷酸化位点 T872。因此,出现了这样一个问题,即 T872 的磷酸化是否参与了受体的下调,以及该位点的磷酸化是否也由 CaMKIIβ 或另一种蛋白激酶介导。在这里,我们表明,GABA 中 T872 的突变失活阻止了培养神经元中受体的降解。我们发现,除了 CaMKIIβ 之外,ERK1/2 也参与了生理和缺血条件下 GABA 受体的降解途径。与我们之前的观点相反,CaMKIIβ 似乎不会直接磷酸化 S867。相反,数据支持这样一种机制,即 CaMKIIβ 激活 ERK1/2,然后 ERK1/2 磷酸化 GABA 中的 S867 和 T872。在将神经元置于缺血应激后阻断 ERK 活性可完全将下调的 GABA 受体表达恢复至正常水平。因此,阻止 ERK1/2 介导的 GABA 中的 S867/T872 磷酸化是抑制缺血应激后受体病理性下调的机会,并有望恢复健康的神经元兴奋/抑制平衡。