Department of Dermatology, the Frist Affiliated Hospital of Xi'an Jiaotong University, 277 West Yanta Road, 710061, Xi'an, Shaanxi, People's Republic of China.
Center for Translational Medicine, the Frist Affiliated Hospital of Xi'an Jiaotong University, 277 West Yanta Road, 710061, Xi'an, Shaanxi, People's Republic of China.
BMC Microbiol. 2023 Sep 22;23(1):265. doi: 10.1186/s12866-023-03020-7.
Vitiligo has been correlated with an abnormal gut microbiota. We aimed to systematically identify characteristics of the gut microbial compositions, genetic functions, and potential metabolic features in patients with non-segmental vitiligo.
Twenty-five patients with non-segmental vitiligo and 25 matched healthy controls (HCs) were enrolled. Metagenomic sequencing and bioinformatic analysis were performed to determine the gut microbiota profiles. Differences in gut microbiota diversity and composition between patients with vitiligo and HCs were analyzed. Gene functions and gut metabolic modules were predicted with the Kyoto Encyclopedia of Gene and Genomes (KEGG) and MetaCyc databases.
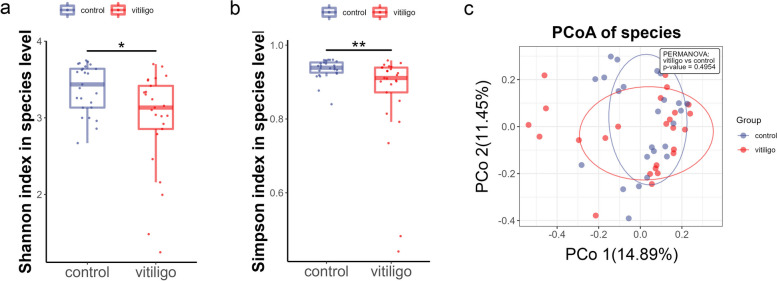
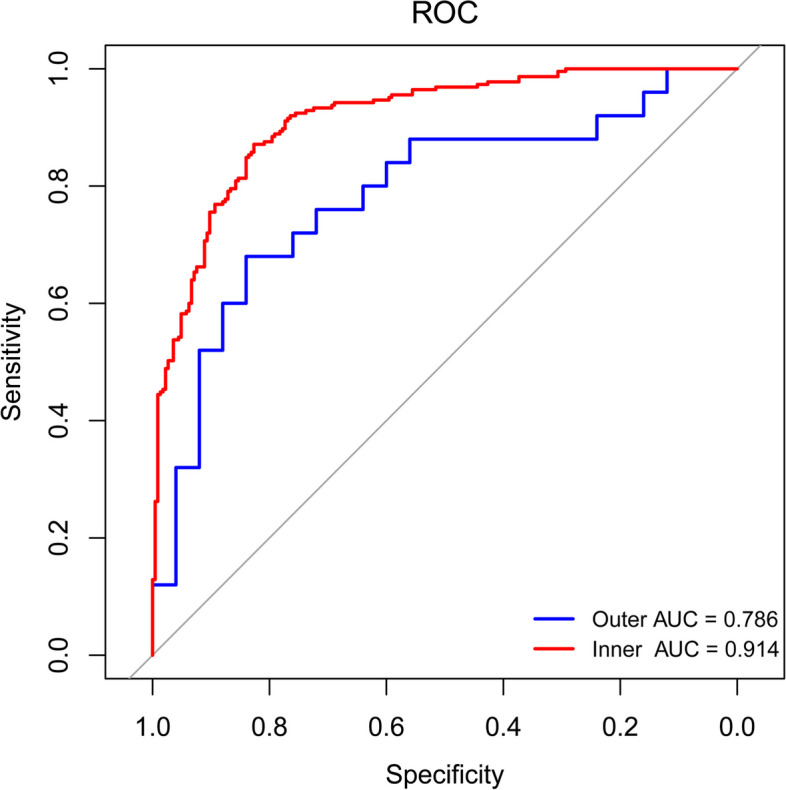
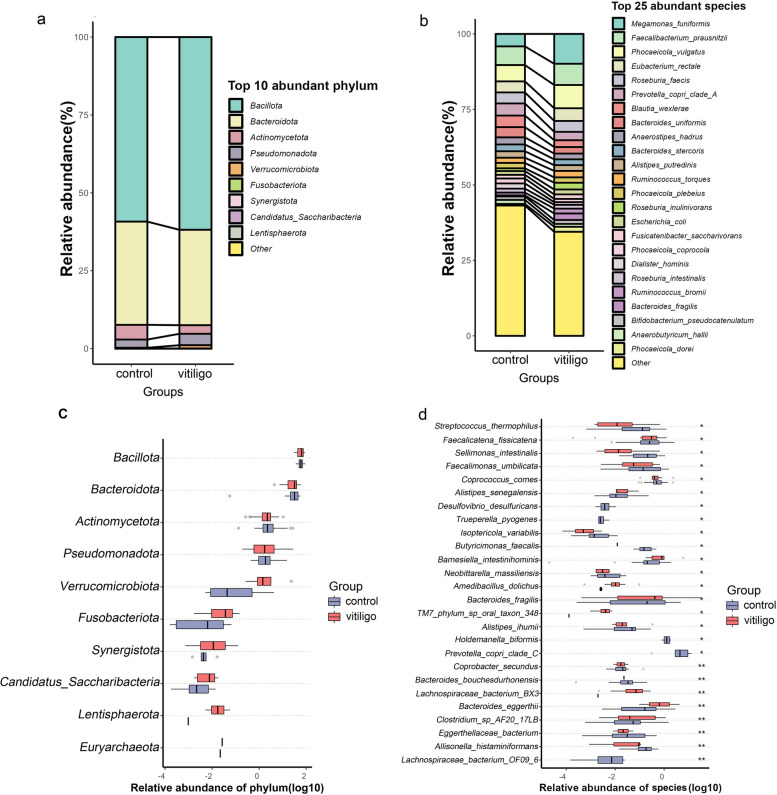
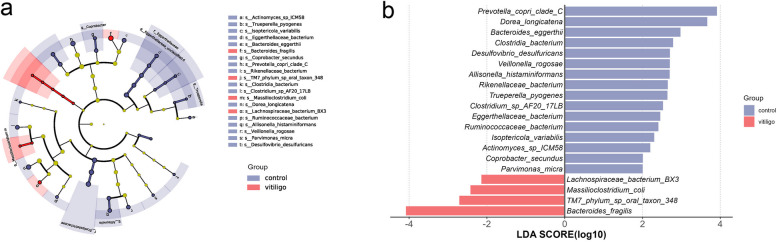
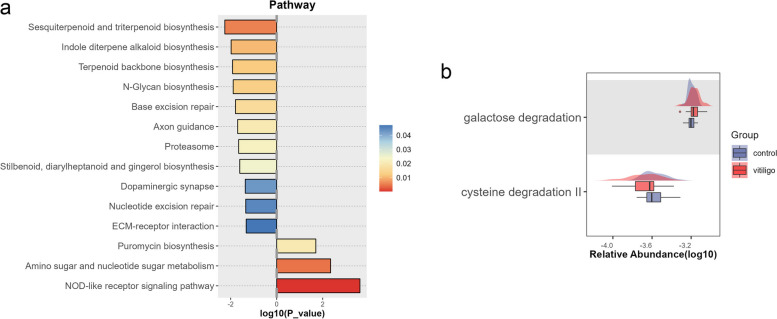
Compared with HCs, alpha diversity of intestinal microbiome in vitiligo patients was significantly reduced. At the species level, the relative abundance of Staphylococcus thermophiles was decreased, and that of Bacteroides fragilis was increased in patients with vitiligo compared with those of the HCs. Linear discriminant analysis (LDA) effect size (LEfSe) analysis revealed representative microbial markers of Lachnospiraceae_bacterium_BX3, Massilioclostridium_coli, TM7_phylum_sp_oral_taxon_348 and Bacteroides_fragilis for patients with vitiligo. KEGG gene function analysis showed that the NOD-like receptor signaling pathway was significantly enriched in patients with vitiligo. Gut metabolic modules (GMMs) analysis showed that cysteine degradation was significantly down-regulated, and galactose degradation was up-regulated in patients with vitiligo. A panel of 28 microbial features was constructed to distinguish patients with vitiligo from HCs.
The gut microbial profiles and genetic functions of patients with vitiligo were distinct from those of the HCs. The identified gut microbial markers may potentially be used for earlier diagnosis and treatment targets.
白癜风与肠道微生物群异常有关。我们旨在系统地识别非节段性白癜风患者的肠道微生物组成、遗传功能和潜在代谢特征。
纳入 25 例非节段性白癜风患者和 25 例匹配的健康对照者(HCs)。进行宏基因组测序和生物信息学分析以确定肠道微生物群谱。分析白癜风患者和 HCs 之间肠道微生物多样性和组成的差异。使用京都基因与基因组百科全书(KEGG)和 MetaCyc 数据库预测基因功能和肠道代谢模块。
与 HCs 相比,白癜风患者肠道微生物组的 alpha 多样性显著降低。在物种水平上,与 HCs 相比,白癜风患者中嗜热链球菌的相对丰度降低,脆弱拟杆菌的相对丰度增加。线性判别分析(LDA)效应量(LEfSe)分析显示,代表微生物标志物的lachnospiraceae_bacterium_BX3、Massilioclostridium_coli、TM7_phylum_sp_oral_taxon_348 和脆弱拟杆菌,用于白癜风患者。KEGG 基因功能分析显示,NOD 样受体信号通路在白癜风患者中显著富集。肠道代谢模块(GMMs)分析显示,胱氨酸降解显著下调,半乳糖降解上调。构建了一组 28 种微生物特征,用于区分白癜风患者和 HCs。
白癜风患者的肠道微生物谱和遗传功能与 HCs 不同。鉴定的肠道微生物标志物可能可用于早期诊断和治疗靶点。