Illarionov Alexey, Sakipov Serzhan, Pereyaslavets Leonid, Kurnikov Igor V, Kamath Ganesh, Butin Oleg, Voronina Ekaterina, Ivahnenko Ilya, Leontyev Igor, Nawrocki Grzegorz, Darkhovskiy Mikhail, Olevanov Michael, Cherniavskyi Yevhen K, Lock Christopher, Greenslade Sean, Sankaranarayanan Subramanian Krs, Kurnikova Maria G, Potoff Jeffrey, Kornberg Roger D, Levitt Michael, Fain Boris
InterX Inc. (a Subsidiary of NeoTX Therapeutics Ltd.), 805 Allston Way, Berkeley, California 94710, United States.
Lomonosov MSU, Skobeltsyn Institute of Nuclear Physics, Moscow, 119991, Russia.
J Am Chem Soc. 2023 Nov 1;145(43):23620-23629. doi: 10.1021/jacs.3c07628. Epub 2023 Oct 19.
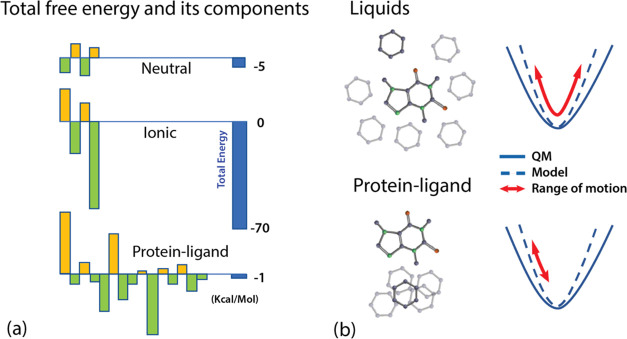
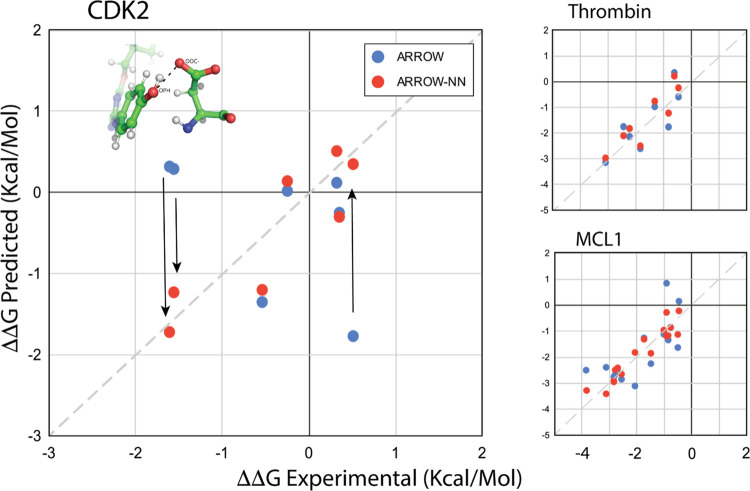
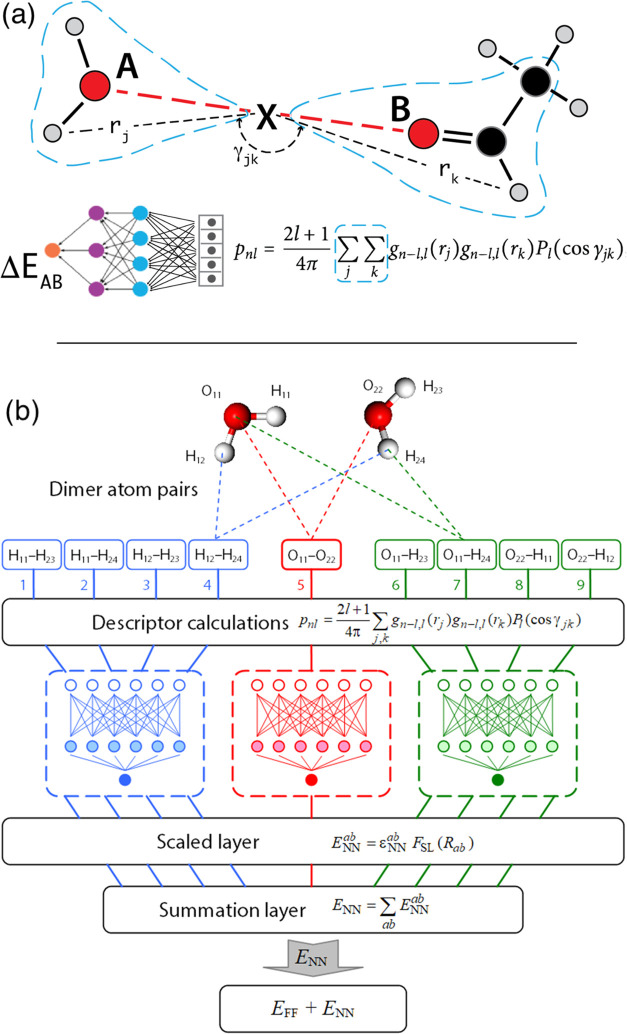
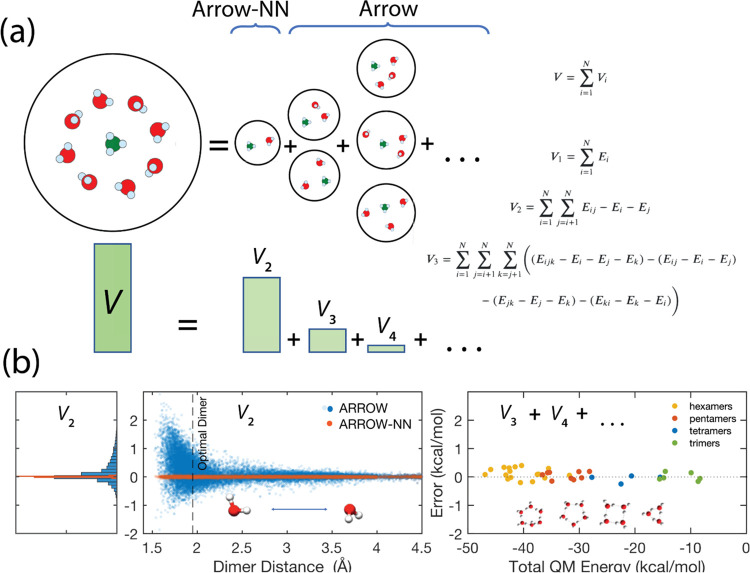
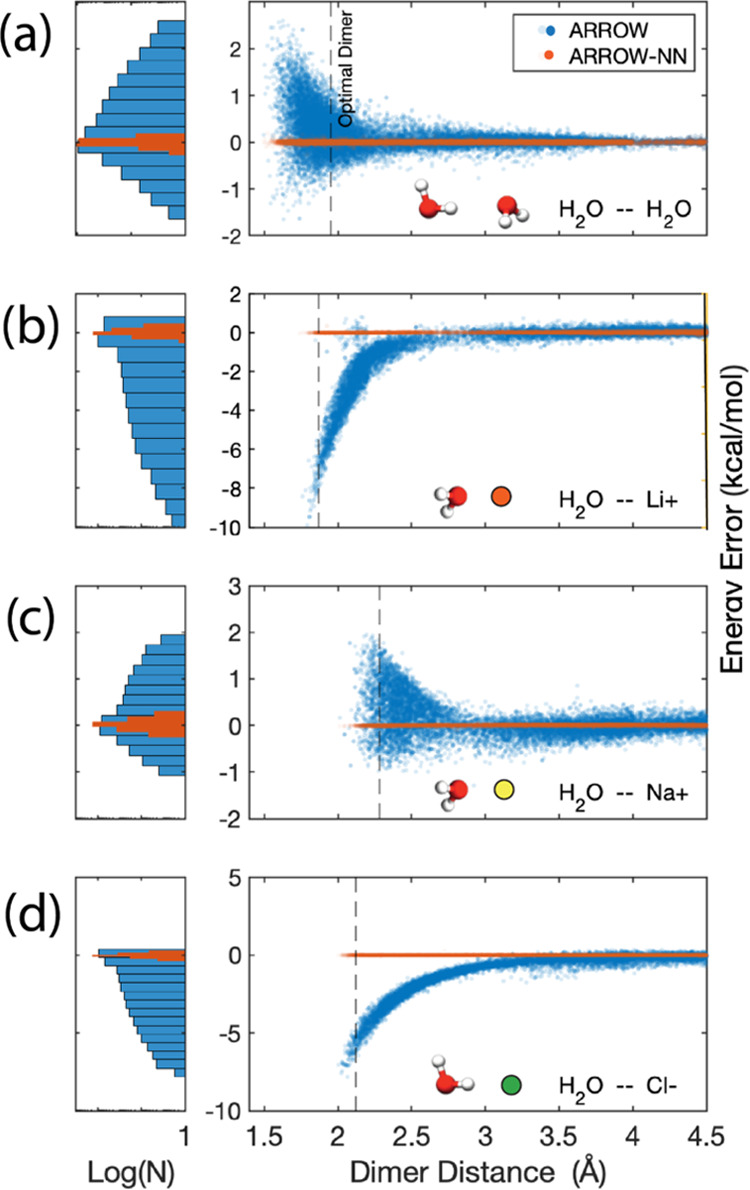
A key goal of molecular modeling is the accurate reproduction of the true quantum mechanical potential energy of arbitrary molecular ensembles with a tractable classical approximation. The challenges are that analytical expressions found in general purpose force fields struggle to faithfully represent the intermolecular quantum potential energy surface at close distances and in strong interaction regimes; that the more accurate neural network approximations do not capture crucial physics concepts, e.g., nonadditive inductive contributions and application of electric fields; and that the ultra-accurate narrowly targeted models have difficulty generalizing to the entire chemical space. We therefore designed a hybrid wide-coverage intermolecular interaction model consisting of an analytically polarizable force field combined with a short-range neural network correction for the total intermolecular interaction energy. Here, we describe the methodology and apply the model to accurately determine the properties of water, the free energy of solvation of neutral and charged molecules, and the binding free energy of ligands to proteins. The correction is subtyped for distinct chemical species to match the underlying force field, to segment and reduce the amount of quantum training data, and to increase accuracy and computational speed. For the systems considered, the hybrid ab initio parametrized Hamiltonian reproduces the two-body dimer quantum mechanics (QM) energies to within 0.03 kcal/mol and the nonadditive many-molecule contributions to within 2%. Simulations of molecular systems using this interaction model run at speeds of several nanoseconds per day.
分子建模的一个关键目标是通过易于处理的经典近似准确再现任意分子系综的真实量子力学势能。挑战在于,通用力场中的解析表达式难以在近距离和强相互作用区域忠实地表示分子间量子势能面;更精确的神经网络近似无法捕捉关键的物理概念,例如非加和诱导贡献和电场的应用;而超精确的窄目标模型难以推广到整个化学空间。因此,我们设计了一种混合的广泛覆盖分子间相互作用模型,该模型由一个可解析极化的力场与一个用于总分子间相互作用能的短程神经网络校正相结合。在这里,我们描述了该方法,并将该模型应用于准确确定水的性质、中性和带电分子的溶剂化自由能以及配体与蛋白质的结合自由能。针对不同的化学物种对校正进行子类型划分,以匹配基础力场,分割并减少量子训练数据量,并提高准确性和计算速度。对于所考虑的系统,混合从头算参数化哈密顿量将两体二聚体量子力学(QM)能量再现到0.03千卡/摩尔以内,非加和多分子贡献再现到2%以内。使用这种相互作用模型对分子系统进行模拟的速度为每天几纳秒。