Jaffrelot Inizan Théo, Plé Thomas, Adjoua Olivier, Ren Pengyu, Gökcan Hatice, Isayev Olexandr, Lagardère Louis, Piquemal Jean-Philip
Sorbonne Université, Laboratoire de Chimie Théorique UMR 7616 CNRS Paris 75005 France
Department of Biomedical Engineering, University of Texas at Austin Austin Texas USA.
Chem Sci. 2023 Apr 4;14(20):5438-5452. doi: 10.1039/d2sc04815a. eCollection 2023 May 24.
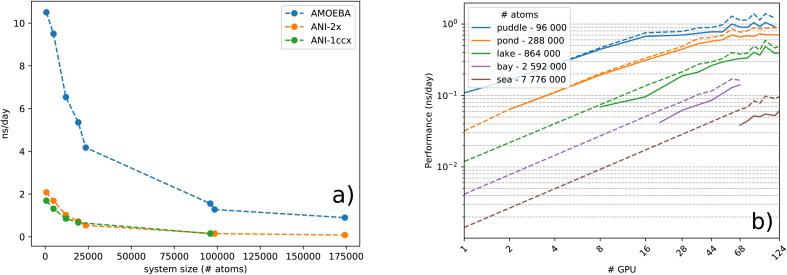
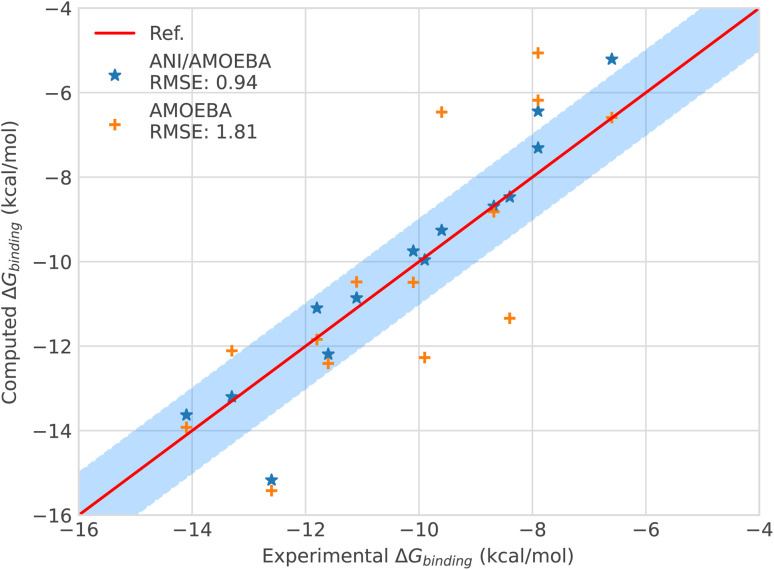
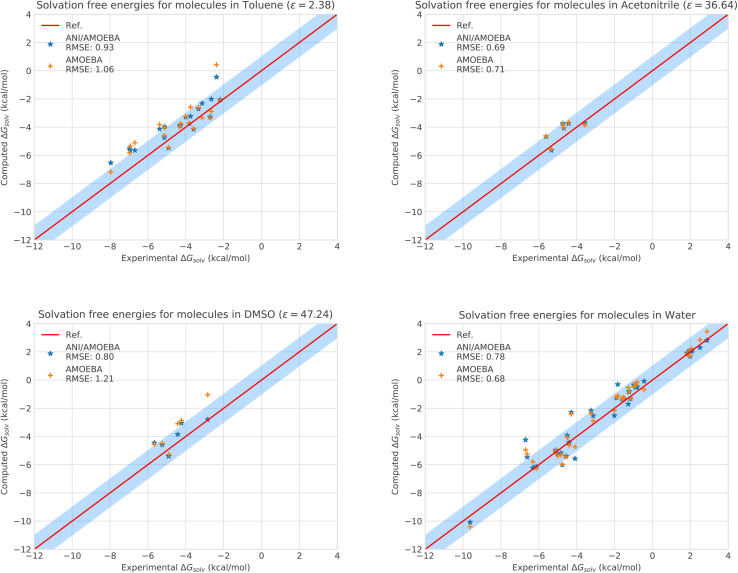
Deep-HP is a scalable extension of the Tinker-HP multi-GPU molecular dynamics (MD) package enabling the use of Pytorch/TensorFlow Deep Neural Network (DNN) models. Deep-HP increases DNNs' MD capabilities by orders of magnitude offering access to ns simulations for 100k-atom biosystems while offering the possibility of coupling DNNs to any classical (FFs) and many-body polarizable (PFFs) force fields. It allows therefore the introduction of the ANI-2X/AMOEBA hybrid polarizable potential designed for ligand binding studies where solvent-solvent and solvent-solute interactions are computed with the AMOEBA PFF while solute-solute ones are computed by the ANI-2X DNN. ANI-2X/AMOEBA explicitly includes AMOEBA's physical long-range interactions an efficient Particle Mesh Ewald implementation while preserving ANI-2X's solute short-range quantum mechanical accuracy. The DNN/PFF partition can be user-defined allowing for hybrid simulations to include key ingredients of biosimulation such as polarizable solvents, polarizable counter ions, … ANI-2X/AMOEBA is accelerated using a multiple-timestep strategy focusing on the model's contributions to low-frequency modes of nuclear forces. It primarily evaluates AMOEBA forces while including ANI-2X ones only correction-steps resulting in an order of magnitude acceleration over standard Velocity Verlet integration. Simulating more than 10 μs, we compute charged/uncharged ligand solvation free energies in 4 solvents, and absolute binding free energies of host-guest complexes from SAMPL challenges. ANI-2X/AMOEBA average errors are discussed in terms of statistical uncertainty and appear in the range of chemical accuracy compared to experiment. The availability of the Deep-HP computational platform opens the path towards large-scale hybrid DNN simulations, at force-field cost, in biophysics and drug discovery.
Deep-HP是Tinker-HP多GPU分子动力学(MD)软件包的可扩展扩展,支持使用Pytorch/TensorFlow深度神经网络(DNN)模型。Deep-HP将DNN的MD能力提高了几个数量级,能够对100k原子的生物系统进行纳秒级模拟,同时还提供了将DNN与任何经典(FFs)和多体极化(PFFs)力场耦合的可能性。因此,它允许引入为配体结合研究设计的ANI-2X/AMOEBA混合极化势,其中溶剂-溶剂和溶剂-溶质相互作用用AMOEBA PFF计算,而溶质-溶质相互作用由ANI-2X DNN计算。ANI-2X/AMOEBA明确包括AMOEBA的物理长程相互作用——一种高效的粒子网格埃瓦尔德实现,同时保留了ANI-2X溶质短程量子力学精度。DNN/PFF分区可以由用户定义,允许混合模拟包含生物模拟的关键要素,如极化溶剂、极化反离子等。ANI-2X/AMOEBA使用多时间步策略加速,该策略侧重于模型对核力低频模式的贡献。它主要评估AMOEBA力,同时仅包括ANI-2X力进行校正步骤,与标准的速度Verlet积分相比,加速了一个数量级。在模拟超过10微秒的情况下,我们计算了4种溶剂中带电/不带电配体的溶剂化自由能,以及来自SAMPL挑战的主客体复合物的绝对结合自由能。ANI-2X/AMOEBA的平均误差根据统计不确定性进行了讨论,与实验相比,其误差在化学精度范围内。Deep-HP计算平台的可用性为生物物理学和药物发现中以力场成本进行大规模混合DNN模拟开辟了道路。