Jiang Lindong, Xu Chao, Bai Yuntong, Liu Anqi, Gong Yun, Wang Yu-Ping, Deng Hong-Wen
Tulane Center of Biomedical Informatics and Genomics, School of Medicine, Tulane University, New Orleans, LA, 70112, USA.
Department of Biostatistics and Epidemiology, University of Oklahoma Health Sciences Center, Oklahoma City, OK, 73104, USA.
NPJ Precis Oncol. 2024 Jan 5;8(1):4. doi: 10.1038/s41698-023-00494-6.
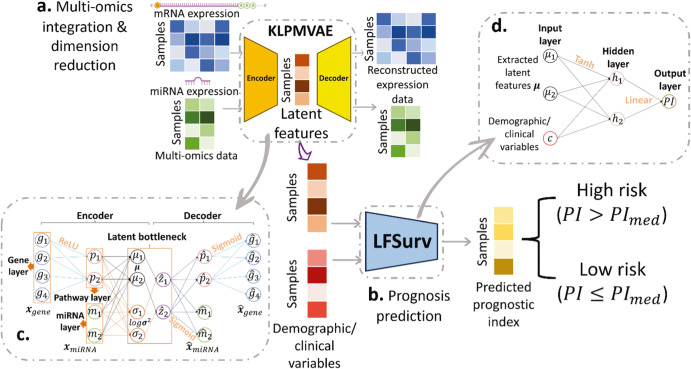
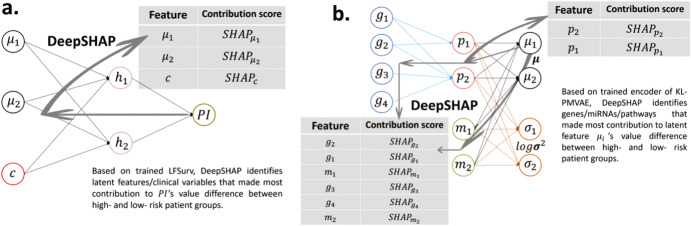
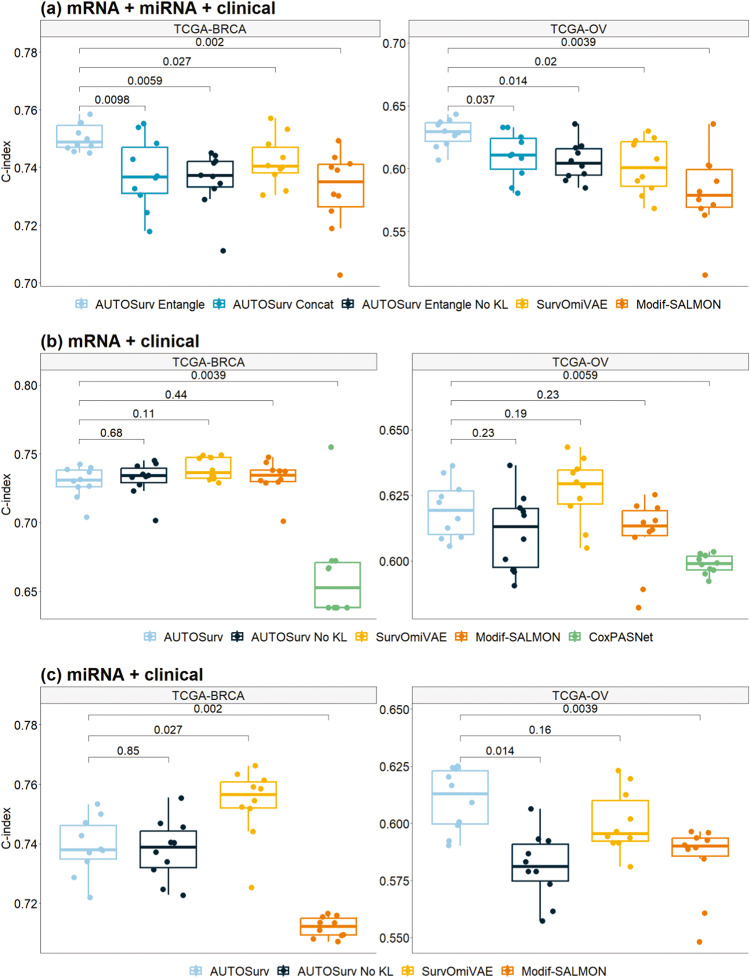
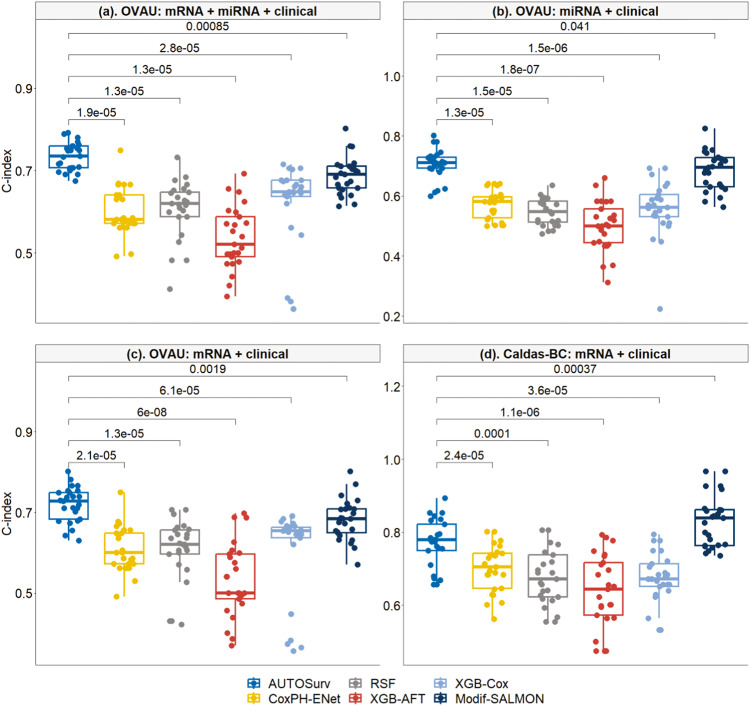
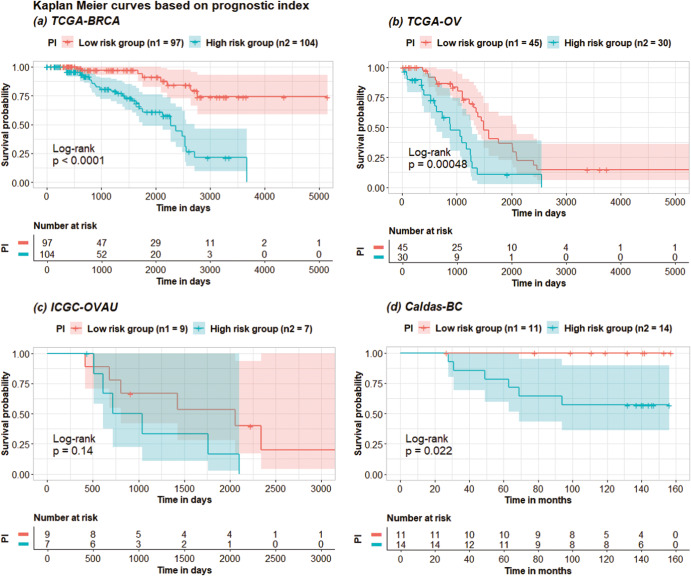
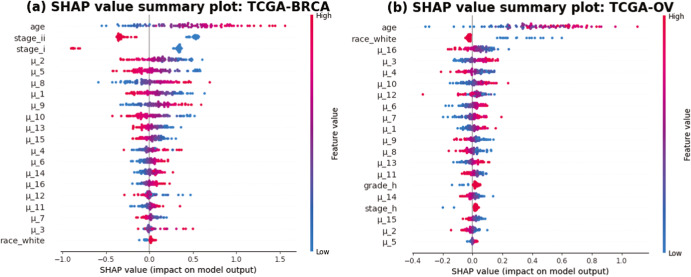
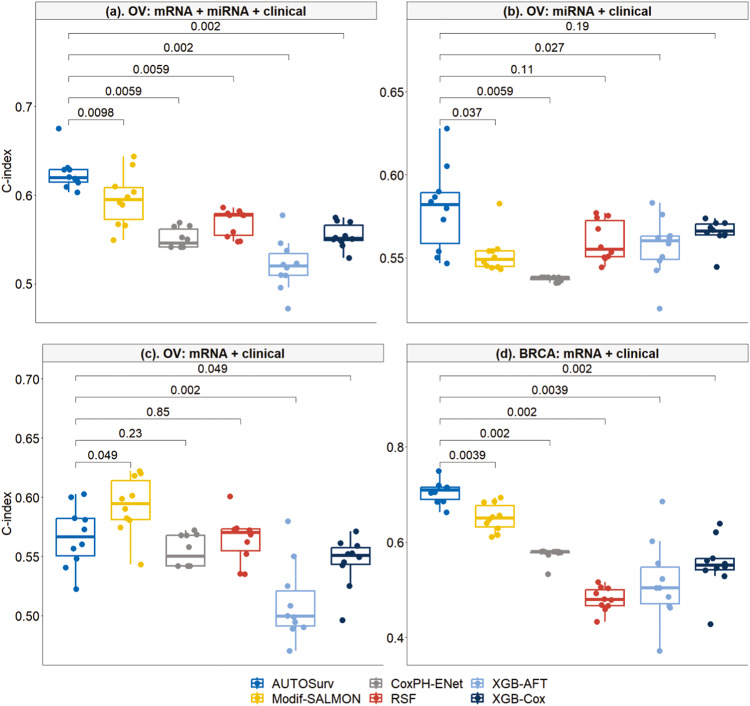
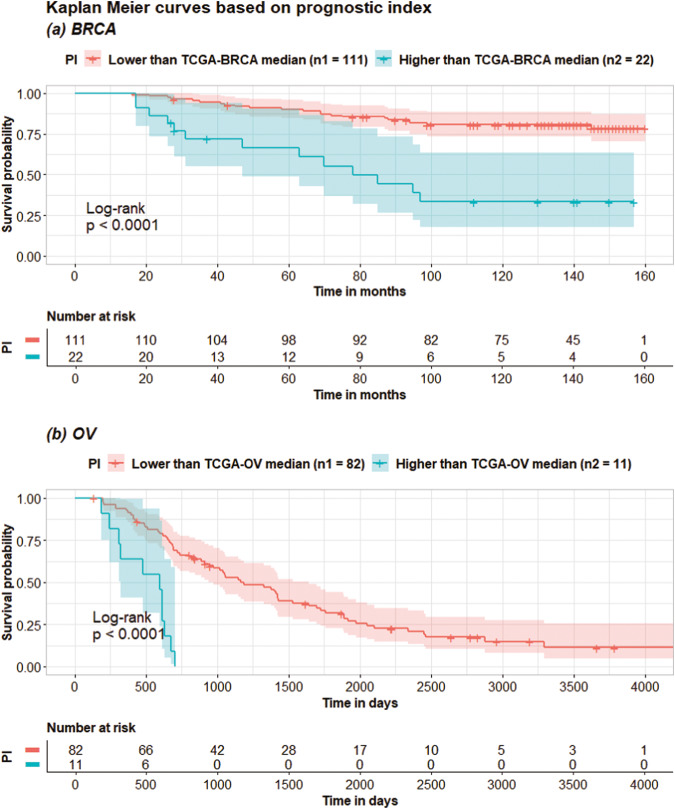
Accurate prognosis for cancer patients can provide critical information for optimizing treatment plans and improving life quality. Combining omics data and demographic/clinical information can offer a more comprehensive view of cancer prognosis than using omics or clinical data alone and can also reveal the underlying disease mechanisms at the molecular level. In this study, we developed and validated a deep learning framework to extract information from high-dimensional gene expression and miRNA expression data and conduct prognosis prediction for breast cancer and ovarian-cancer patients using multiple independent multi-omics datasets. Our model achieved significantly better prognosis prediction than the current machine learning and deep learning approaches in various settings. Moreover, an interpretation method was applied to tackle the "black-box" nature of deep neural networks and we identified features (i.e., genes, miRNA, demographic/clinical variables) that were important to distinguish predicted high- and low-risk patients. The significance of the identified features was partially supported by previous studies.
准确预测癌症患者的预后可为优化治疗方案和提高生活质量提供关键信息。与单独使用组学或临床数据相比,整合组学数据与人口统计学/临床信息能提供更全面的癌症预后观点,还能在分子水平揭示潜在的疾病机制。在本研究中,我们开发并验证了一个深度学习框架,用于从高维基因表达和miRNA表达数据中提取信息,并使用多个独立的多组学数据集对乳腺癌和卵巢癌患者进行预后预测。在各种情况下,我们的模型在预后预测方面显著优于当前的机器学习和深度学习方法。此外,我们应用了一种解释方法来解决深度神经网络的“黑箱”性质问题,并确定了区分预测的高风险和低风险患者的重要特征(即基因、miRNA、人口统计学/临床变量)。先前的研究部分支持了所确定特征的重要性。