Adams Jason S, Chen Haoyu, Ricciardulli Tomas, Vijayaraghavan Sucharita, Sampath Abinaya, Flaherty David W
Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Champaign, Illinois 61801, United States.
School of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, United States.
ACS Catal. 2024 Feb 15;14(5):3248-3265. doi: 10.1021/acscatal.3c05072. eCollection 2024 Mar 1.
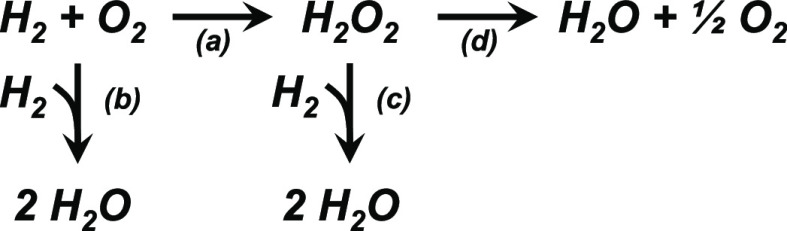
Au nanoparticles catalyze the activation and conversion of small molecules with rates and kinetic barriers that depend on the dimensions of the nanoparticle, composition of the support, and presence of catalytically culpable water molecules that solvate these interfaces. Here, molecular interpretations of steady-state rate measurements, kinetic isotope effects, and structural characterizations reveal how the interface of Au nanoparticles, liquid water, and metal oxide supports mediate the kinetically relevant activation of H and sequential reduction of O-derived intermediates during the formation of HO and HO. Rates of H consumption are 10-100 fold greater on Au nanoparticles supported on metal oxides (e.g., titania) compared to more inert and hydrophobic materials (carbon, boron nitride). Similarly, Au nanoparticles on reducible and Lewis acidic supports (e.g., lanthana) bind dioxygen intermediates more strongly and present lower barriers (<22 kJ mol) for O-O bond dissociation than inert interfaces formed with silica (>70 kJ mol). Selectivities for HO formation increase significantly as the diameters of the Au nanoparticles increase because differences in nanoparticle size change the relative fractions of exposed sites that exist at Au-support interfaces. In contrast, site-normalized rates and barriers for H activation depend weakly on the size of Au nanoparticles and the associated differences in active site motifs. These findings suggest that HO aids the activation of H at sites present across all surface Au atoms when nanoparticles are solvated by water. However, molecular O preferentially binds and dissociates at Au-support interfaces, leading to greater structure sensitivity for barriers of O-O dissociation across different support identities and sizes of Au nanoparticles. These insights differ from prior knowledge from studies of gas-phase reactions of H and O upon Au nanoparticle catalysts within dilute vapor pressures of water (10 to 0.1 kPa HO), in which catalysis occurs at the perimeter of the Au-support interface. In contrast, contacting Au catalysts with liquid water (55.5 M HO) expands catalysis to all surface Au atoms and enables appreciable HO formation.
金纳米颗粒催化小分子的活化和转化,其速率和动力学势垒取决于纳米颗粒的尺寸、载体的组成以及溶剂化这些界面的具有催化作用的水分子的存在。在此,对稳态速率测量、动力学同位素效应和结构表征的分子解释揭示了金纳米颗粒、液态水和金属氧化物载体的界面如何在形成HO和HO的过程中介导与动力学相关的H活化以及O衍生中间体的顺序还原。与更惰性和疏水性的材料(碳、氮化硼)相比,负载在金属氧化物(如二氧化钛)上的金纳米颗粒上的H消耗速率要高10 - 100倍。同样,负载在可还原和路易斯酸性载体(如氧化镧)上的金纳米颗粒与双氧中间体的结合更强,并且与由二氧化硅形成的惰性界面(>70 kJ/mol)相比,O - O键解离的势垒更低(<22 kJ/mol)。随着金纳米颗粒直径的增加,HO形成的选择性显著增加,因为纳米颗粒尺寸的差异改变了金 - 载体界面处暴露位点的相对比例。相比之下,H活化的位点归一化速率和势垒对金纳米颗粒的尺寸以及活性位点基序的相关差异的依赖性较弱。这些发现表明,当纳米颗粒被水溶剂化时,HO有助于在所有表面金原子存在的位点上活化H。然而,分子O优先在金 - 载体界面结合和解离,导致不同载体特性和金纳米颗粒尺寸的O - O解离势垒具有更高的结构敏感性。这些见解不同于在水的稀蒸气压(10至0.1 kPa HO)下金纳米颗粒催化剂上H和O的气相反应研究的先验知识,在该研究中催化作用发生在金 - 载体界面的周边。相比之下,使金催化剂与液态水(55.5 M HO)接触将催化作用扩展到所有表面金原子,并能够形成可观的HO。