Siriraj Center of Research Excellence in Metabolomics and Systems Biology (SiCORE-MSB), Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand.
Siriraj Metabolomics and Phenomics Center, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand.
Gigascience. 2024 Jan 2;13. doi: 10.1093/gigascience/giae005.
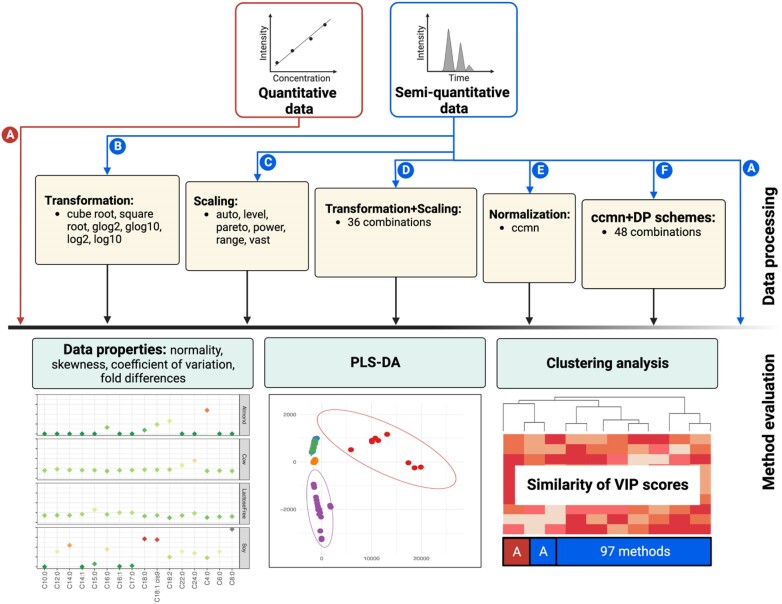
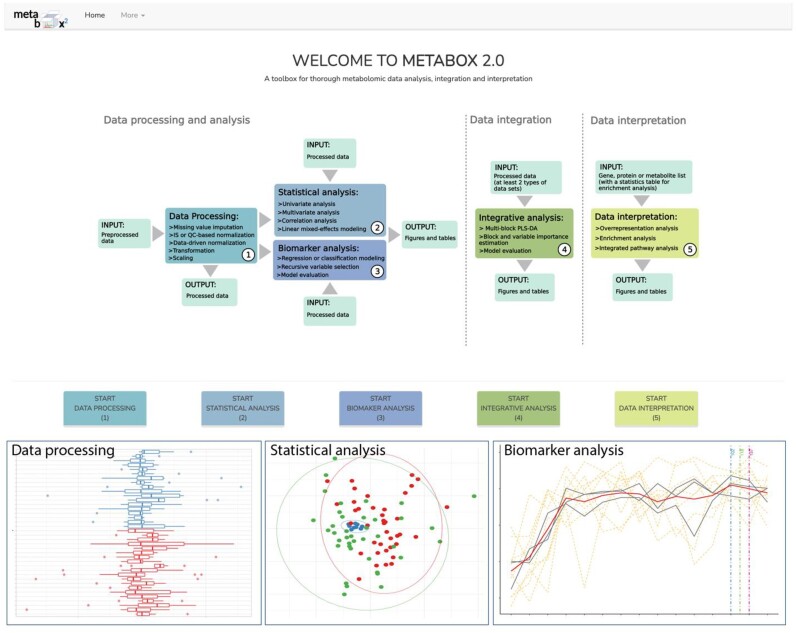
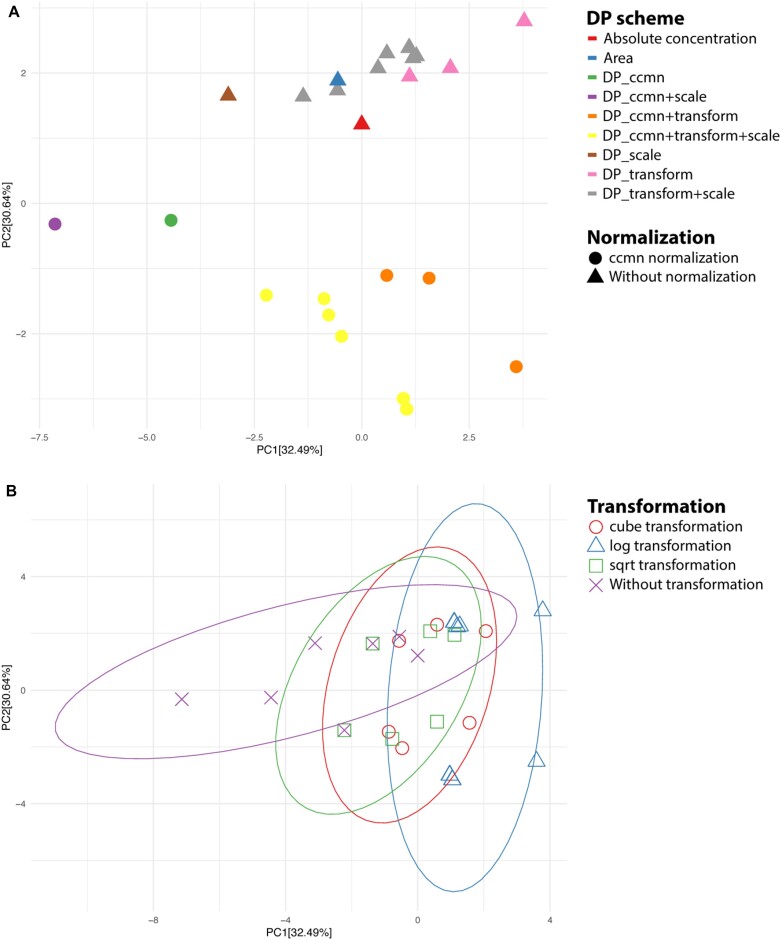
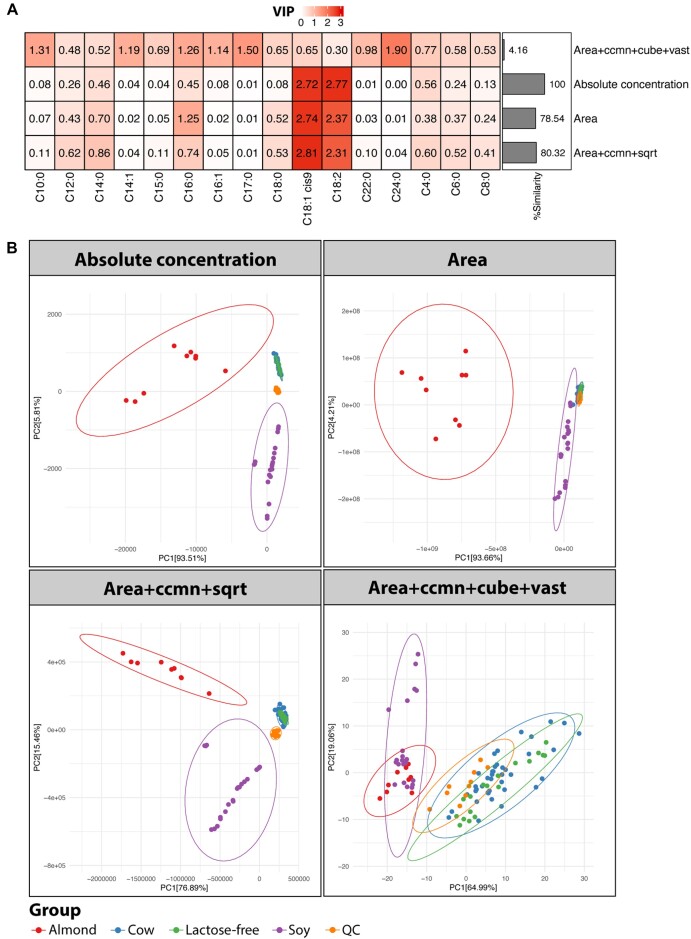
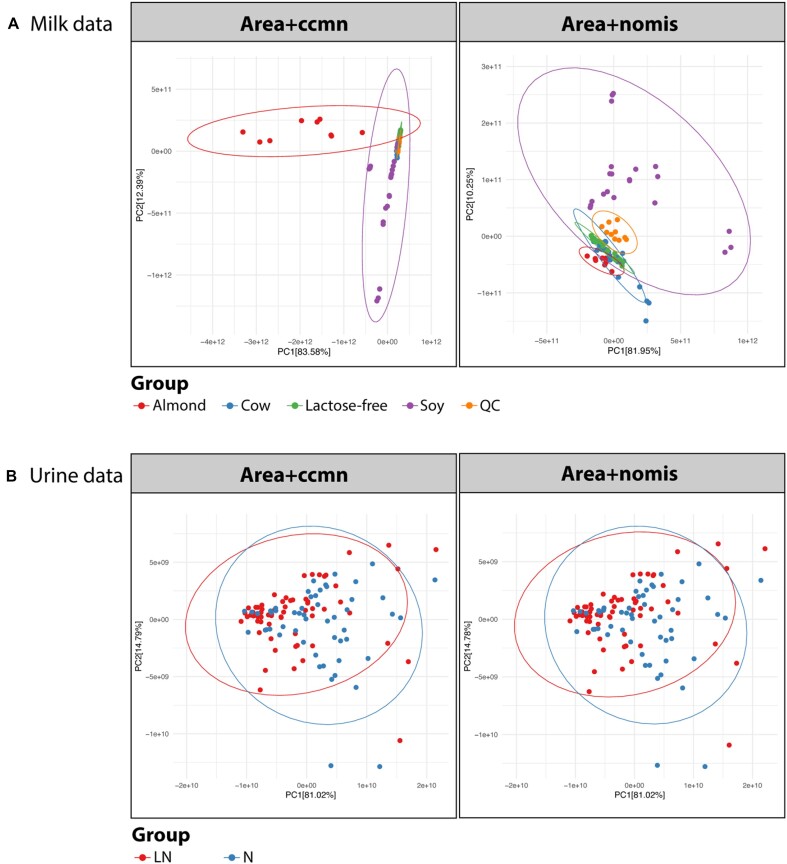
In classic semiquantitative metabolomics, metabolite intensities are affected by biological factors and other unwanted variations. A systematic evaluation of the data processing methods is crucial to identify adequate processing procedures for a given experimental setup. Current comparative studies are mostly focused on peak area data but not on absolute concentrations. In this study, we evaluated data processing methods to produce outputs that were most similar to the corresponding absolute quantified data. We examined the data distribution characteristics, fold difference patterns between 2 metabolites, and sample variance. We used 2 metabolomic datasets from a retail milk study and a lupus nephritis cohort as test cases. When studying the impact of data normalization, transformation, scaling, and combinations of these methods, we found that the cross-contribution compensating multiple standard normalization (ccmn) method, followed by square root data transformation, was most appropriate for a well-controlled study such as the milk study dataset. Regarding the lupus nephritis cohort study, only ccmn normalization could slightly improve the data quality of the noisy cohort. Since the assessment accounted for the resemblance between processed data and the corresponding absolute quantified data, our results denote a helpful guideline for processing metabolomic datasets within a similar context (food and clinical metabolomics). Finally, we introduce Metabox 2.0, which enables thorough analysis of metabolomic data, including data processing, biomarker analysis, integrative analysis, and data interpretation. It was successfully used to process and analyze the data in this study. An online web version is available at http://metsysbio.com/metabox.
在经典的半定量代谢组学中,代谢物强度会受到生物因素和其他非期望变化的影响。系统评估数据处理方法对于确定给定实验设置的适当处理程序至关重要。当前的比较研究主要集中在峰面积数据上,而不是绝对浓度。在这项研究中,我们评估了数据处理方法,以生成与相应绝对定量数据最相似的输出。我们检查了数据分布特征、两个代谢物之间的倍数差异模式和样本方差。我们使用了来自零售牛奶研究和狼疮肾炎队列的两个代谢组学数据集作为测试案例。在研究数据归一化、转换、缩放以及这些方法的组合的影响时,我们发现交叉贡献补偿多标准归一化(ccmn)方法,其次是平方根数据转换,最适合像牛奶研究数据集这样的受控研究。关于狼疮肾炎队列研究,只有 ccmn 归一化才能稍微改善嘈杂队列的数据质量。由于评估考虑了处理后数据与相应绝对定量数据之间的相似性,因此我们的结果为在类似背景(食品和临床代谢组学)下处理代谢组学数据集提供了有用的指导。最后,我们介绍了 Metabox 2.0,它可以对代谢组学数据进行全面分析,包括数据处理、生物标志物分析、综合分析和数据解释。它成功地用于处理和分析本研究中的数据。在线网络版本可在 http://metsysbio.com/metabox 获得。