Acellera Labs, C/Doctor Trueta 183, Barcelona 08005, Spain.
Computational Science Laboratory, Universitat Pompeu Fabra, PRBB, C/Doctor Aiguader 88, Barcelona 08003, Spain.
J Chem Inf Model. 2023 Sep 25;63(18):5701-5708. doi: 10.1021/acs.jcim.3c00773. Epub 2023 Sep 11.
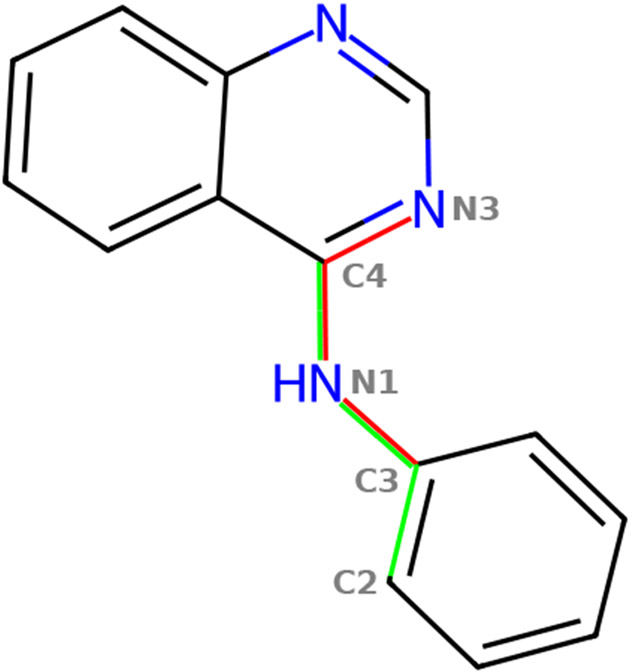
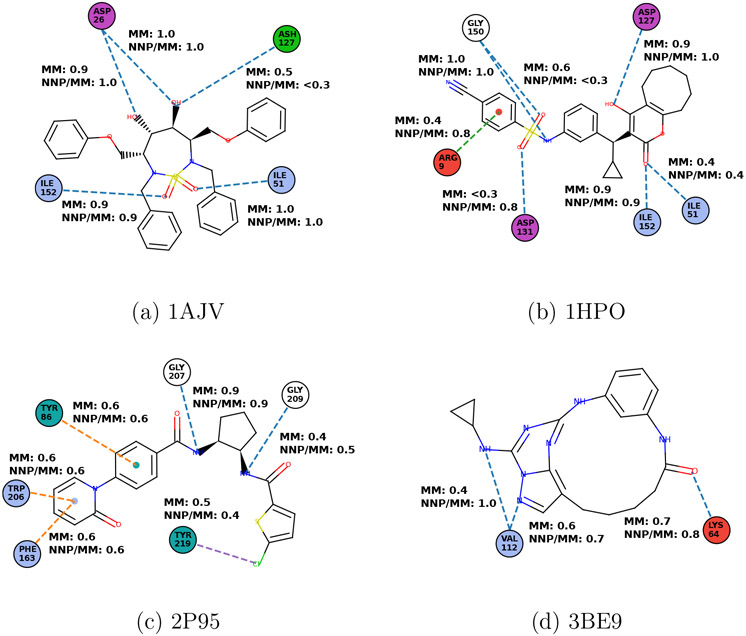
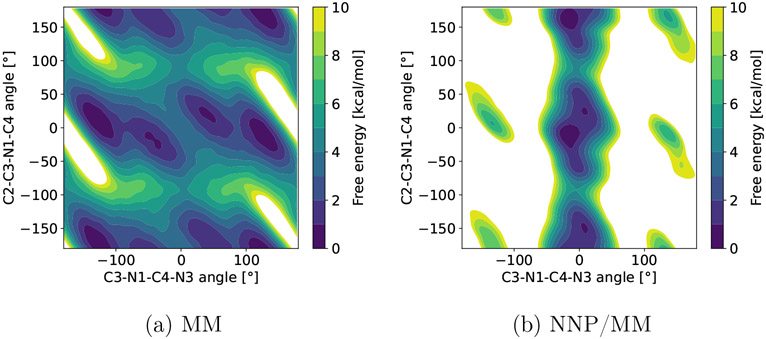
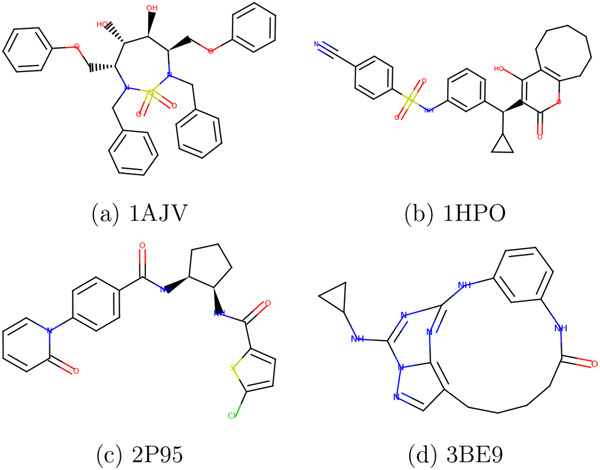
Machine learning potentials have emerged as a means to enhance the accuracy of biomolecular simulations. However, their application is constrained by the significant computational cost arising from the vast number of parameters compared with traditional molecular mechanics. To tackle this issue, we introduce an optimized implementation of the hybrid method (NNP/MM), which combines a neural network potential (NNP) and molecular mechanics (MM). This approach models a portion of the system, such as a small molecule, using NNP while employing MM for the remaining system to boost efficiency. By conducting molecular dynamics (MD) simulations on various protein-ligand complexes and metadynamics (MTD) simulations on a ligand, we showcase the capabilities of our implementation of NNP/MM. It has enabled us to increase the simulation speed by ∼5 times and achieve a combined sampling of 1 μs for each complex, marking the longest simulations ever reported for this class of simulations.
机器学习方法已经成为提高生物分子模拟准确性的一种手段。然而,由于与传统分子力学相比,它们的应用受到大量参数带来的巨大计算成本的限制。为了解决这个问题,我们引入了一种混合方法(NNP/MM)的优化实现,该方法结合了神经网络势(NNP)和分子力学(MM)。这种方法使用 NNP 来模拟系统的一部分,例如小分子,而对其余系统使用 MM 以提高效率。通过对各种蛋白质-配体复合物进行分子动力学(MD)模拟和对配体进行元动力学(MTD)模拟,我们展示了我们的 NNP/MM 实现的能力。它使我们能够将模拟速度提高约 5 倍,并为每个复合物实现 1μs 的组合采样,这标志着此类模拟中报告的最长模拟。