Departamento de Fisica, Universidade Federal de Santa Catarina, Florianopolis 88040-970, Brasil.
Department of Physiology and Biophysics, Weill Cornell Medical College of Cornell University, New York, New York 10065, United States.
J Chem Theory Comput. 2024 Aug 13;20(15):6518-6530. doi: 10.1021/acs.jctc.4c00205. Epub 2024 Aug 1.
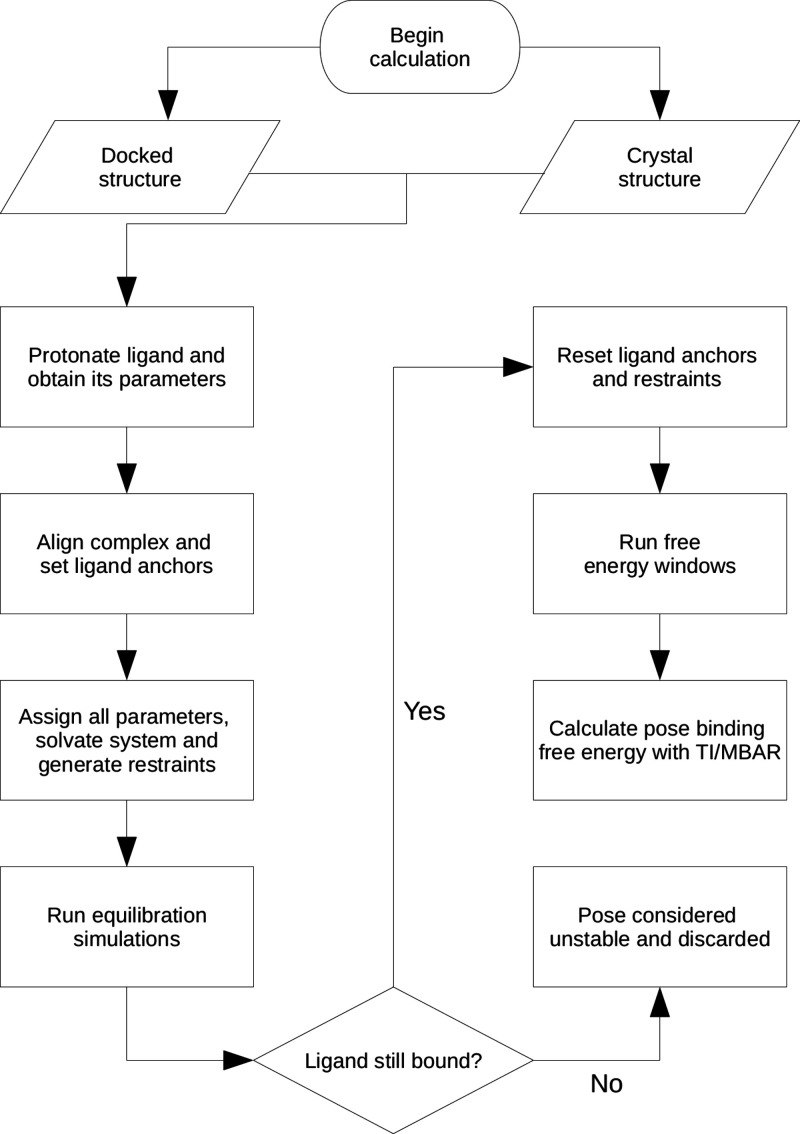
Absolute binding free energy (ABFE) calculations with all-atom molecular dynamics (MD) have the potential to greatly reduce costs in the first stages of drug discovery. Here, we introduce BAT2, the new version of the Binding Affinity Tool (BAT.py), designed to combine full automation of ABFE calculations with high-performance MD simulations, making it a potential tool for virtual screening. We describe and test several changes and new features that were incorporated into the code, such as relative restraints between the protein and the ligand instead of using fixed dummy atoms, support for the OpenMM simulation engine, a merged approach to the application/release of restraints, support for cobinders and proteins with multiple chains, and many others. We also reduced the simulation times for each ABFE calculation, assessing the effect on the expected robustness and accuracy of the calculations.
使用全原子分子动力学 (MD) 进行绝对结合自由能 (ABFE) 计算有可能大大降低药物发现的早期成本。在这里,我们引入了 BAT2,即 Binding Affinity Tool (BAT.py) 的新版本,旨在将 ABFE 计算的完全自动化与高性能 MD 模拟相结合,使其成为虚拟筛选的潜在工具。我们描述并测试了代码中合并的几个更改和新功能,例如,在不使用固定虚拟原子的情况下,在蛋白质和配体之间使用相对约束,支持 OpenMM 模拟引擎,合并应用/释放约束的方法,支持 cobinders 和具有多个链的蛋白质,以及其他许多功能。我们还减少了每个 ABFE 计算的模拟时间,评估了对计算的预期稳健性和准确性的影响。